Our look is new. Our purpose is the same. Learn more about our brand evolution.
Our look is new. Our purpose is the same. Learn more about our brand evolution.
Facioscapulohumeral muscular dystrophy has been linked to two distinct genetic mechanisms. The most common, found in 95 percent or patients, is called FSHD Type 1, or FSHD1. The remaining 5 percent is called FSHD Type 2 (FSHD2), which is linked to mutations in the genes SMCHD1, DNMT3B, and LIRF1. Additional genetic factors are an active area of research. See also Genetic Testing.
Facioscapulohumeral muscular dystrophy Type 1 (also called FSHD1A or FSHMD1A) is the most common form of FSHD, accounting for 95 percent of cases. A conservative estimate of incidence is one in 14,000 births throughout the world. However, due to increased experience with FSHD, population-based research, and improved genetic testing, this estimate may be low. A 2014 study in the Netherlands indicates a higher prevalence one in 8,333, and this may still be an undercount due to underdiagnosis.
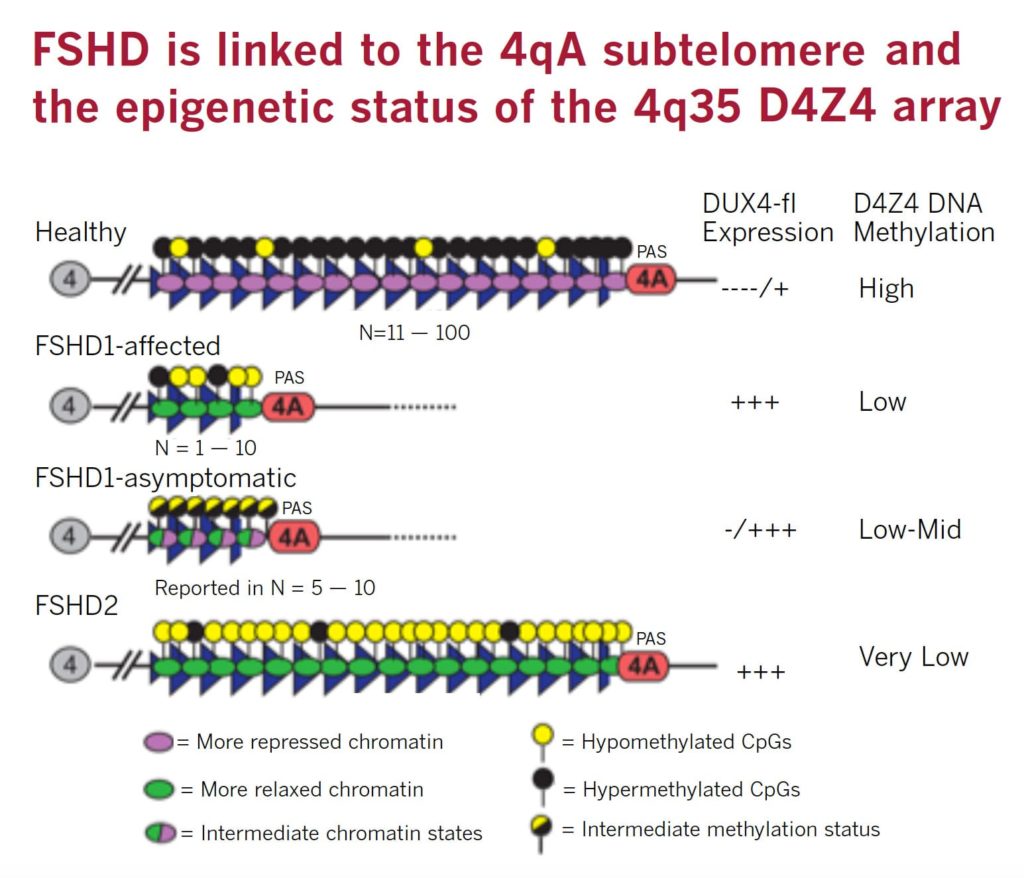
FSHD is caused by ectopic expression of the germline transcription factor DUX4 gene in muscle cells. FSHD1 is associated with the deletion of a chromosomal tandem repeat called D4Z4 near the end of chromosome 4 at the 4q35 location. The D4Z4 region is a polymorphic variable number tandem repeat (VNTR) array consisting of 3.3 kilobase units, and each unit encodes for the DUX4 gene. Unaffected individuals have a chromosome 4 D4Z4 array that has a span of 11 to 150 contiguous units. In individuals with FSHD, this D4Z4 repeat array is contracted to a range between one to 10 contiguous units. The contraction of the D4Z4 repeat array causes hypomethylation and relaxation of chromatin, which activates expression of the toxic DUX4 gene.
The contraction of the D4Z4 repeat array on chromosome 4q35 by itself is not sufficient to cause FSHD. Adjacent to the D4Z4 region, toward the distal end of the chromosome, lies a polyadenylation site that is part of the DUX4 gene and is required for stable expression of the gene in the most distal D4Z4 unit. Chromosome 4 comes in two alleles, called type 4qA and 4qB based on DNA variations distal to the D4Z4 repeat array. Only the 4qA variant in combination with the shortened D4Z4 repeat region is associated with FSHD. Individuals who have a shortened D4Z4 array on a 4qB allele are unaffected due to the absence of the DUX4 polyadenylation site. The D4Z4 repeat array is also present on chromosome 10, but a shortened D4Z4 repeat on chromosome 10 does not cause FSHD because of mutations in the polyadenylation site.
The online database Online Mendelian Inheritance in Man (OMIM) is an excellent source for further information on the science and medicine of FSHD1.
This New York Times article describes the landmark discovery of the genetic mechanism of FSHD: Reanimated ‘Junk’ DNA Is Found to Cause Disease.
Facioscapulohumeral muscular dystrophy Type 2 (also called FSHD1B or FSHMD1B) is much rarer than Type 1 and is thought to account for the majority of the 5 percent of FSHD cases that test negative for Type 1.
FSHD2 is clinically identical to FSHD1 but genetically distinct. In FSHD1, the tandem repeats called D4Z4 on chromosome 4 are deleted, whereas in FSHD2, the number of D4Z4 repeats is in the normal range. Another way of saying this is that there is no “contraction” in D4Z4 at 4q35.
Eighty percent of FSHD2 is caused by the inheritance of two independent genetic variations: a heterozygous loss-of-function mutation in the encoding Structural Maintenance of Chromosomes flexible Hinge Domain containing 1 (SMCHD1) gene combined with the 4qA allele carrying the DUX4 polyadenylation site, making this allele permissive to expression of the toxic DUX4 gene.
More than 52 mutations associated with FSHD have been reported as of early 2014 in SMCHD1. These mutations result in hypomethylation and relaxation of chromatin in all D4Z4 repeat arrays on chromosomes 4 and 10, just as deletions of D4Z4 repeats do. In the presence of the 4qA allele, this is thought to create a permissive environment for the DUX4 gene to be expressed. However, in combination with two non-permissive 4qB alleles, SMCHD1 mutations will not cause FSHD. In other words, FSHD2 is a digenic disease. About 20 percent of FSHD2 individuals with hypomethylation at D4Z4, a SMCHD1 mutation, and a permissive D4Z4 haplotype were asymptomatic, indicating an incomplete penetrance.
Some FSHD families have been identified with an FSHD1 mutation as well as an FSHD2 mutation. Individuals who carry both mutations were shown to be more affected than family members with only one of the two mutations, showing that SMCHD1 can act as a modifier in FSHD1 families. Due to the digenic inheritance pattern and incomplete penetrance, FSHD2 was previously incorrectly referred to as non-chromosome 4-linked FSHD.
The online database Online Mendelian Inheritance in Man (OMIM) is an excellent source for further information on the science and medicine of FSHD2.

