
Since 1998, the FSHD Society has funded groundbreaking research on facioscapulohumeral muscular dystrophy. The Society's grants have contributed to the discovery of the genetic basis of FSHD, shed light on how patients are affected, and unraveled biological processes that lead to muscle weakness and degeneration. They have found targets for potential therapies, developed cellular and animal models for drug discovery, and built tools for clinical trials.
Our grant program invites proposals from researchers around the world. The proposals are reviewed for quality and potential impact by our world-class scientific advisory board. Funded researchers must provide progress reports every six months, and the materials developed during the research must be available to the scientific community. We ensure that our research program is accountable to the FSHD community, whose generous, committed support makes it all possible.

"Without the FSHD Society’s initial belief in me and my lab’s ideas, I probably wouldn’t be working on FSHD now."
Scott Q. Harper, PhD
2023 Awards
Deciphering proteomic signatures of sexual dimorphism in FSHD. Yusuke Nishimura, Liverpool John Moores University. $71,426.
Men with FSHD often experience muscle weakness at a younger age than women, but the reasons for this difference are currently unknown. Female sex hormones may offer some protection against FSHD, but our recent studies found muscle cells from male FSHD patients have more severe disruptions to muscle proteins than cells from female patients even when sex hormones were absent. This project will further investigate sex differences in FSHD using a new mouse model, known as FLExDUX4. Like human patients, male FLExDUX4 mice have more severe muscle pathology than female animals. Also, we recently found that female FlexDUX4 mice respond more positively to exercise training than males.
This project will produce the first comprehensive analysis of thousands of muscle proteins in the FLExDUX4 model, including the effects of exercise training in both male and female animals. We hope our work will generate a better understanding of sex differences in FSHD that could lead to new therapies, e.g. by targeting the processes of FSHD that are specific to either men or women. In addition, when we publish our work, we will make our raw data freely and publicly available to the benefit of the FSHD research community.
Verified all-atom structures of the DUX4-P300 and DUX4-SIX1 interactions. Nir Kalisman, The Hebrew University of Jerusalem. $174,700.
Obtaining all-atom structures of DUX4 in interaction with other proteins by standard structural biology approaches is challenging because of its highly flexible nature. We have previously developed a combined experimental/computational workflow that takes advantage of the recent advances in deep-learning algorithms, and were successful in obtaining structures for two DUX4 interactions.
We propose to apply the same workflow to elucidate the structural basis for two critical interactions: DUX4-P300 and DUX4-SIX1. P300 interacts with DUX4 as the first step towards gene dysregulation and cell toxicity. SIX1 is a regulating factor of myogenesis, and its interaction with DUX4 may disrupt its normal functions. Therefore, successful completion of this proposal will advance our understanding of early steps in DUX4 toxicity by providing the solid base of all-atom structures for key interactions. Such structures are an absolute requisite for the subsequent rational design of small-molecules and peptide therapies that aim to disrupt them.
A novel therapeutic perspective for FSHD muscular dystrophy. Emanuele Mocciaro, San Raffaele Scientific Institute. $159,303.
Facioscapulohumeral muscular dystrophy (FSHD) is the most prevalent progressive myopathy that afflicts both children and adults regardless of the gender. Unfortunately, despite life expectance is not modified, affected individuals eventually requires a wheelchair and no cure or therapeutic option currently available to patients. There is a consensus in the FSHD field indicating the aberrant re-activation of the transcription factor DUX4 as leading cause of the disease. Given its pivotal role in FSHD, blocking DUX4 expression with small molecule drugs is an attractive solution.
In our studies we identified a novel regulator of DUX4 expression that when is blocked significantly decreases DUX4 expression and reduces its toxicity effects. Intriguingly, several drug inhibitors of the novel DUX4 regulator are commercially available and are currently in clinical trial for other diseases representing a big advantage for developing a therapeutic strategy for FSHD. Thanks to our promising preliminary data, we propose to use these small molecule inhibitors and test safety and efficacy in relevant models of FSHD.
This project will allow us to study the potential of inhibiting the toxic expression of DUX4 as an innovative therapeutic approach to treat FSHD.
Pharmacological prevention of DUX4-mediated myonecrosis in vitro & in vivo. Maximilien Buncze. $94,500.
Project description forthcoming.
2022 awards
The application of Whole Exome Sequencing (WES) and methylation levels assessment in the present project will be addressed to provide a molecular diagnosis of FSHD in patients with the clinical signs of disease. Importantly, the project will be addressed to assess the clinical utility of performing WES in FSHD patients with variable D4Z4 RU and epigenetic status. Indeed, the application of WES is expected to improve the molecular diagnosis and facilitate the detection of novel disease genes, pathogenic variants in known genes (SMCHD1, DNMT3B, LRIF1) and low-level mosaicism associated with FSHD phenotypes.
The present study could also allow testing the possible use of methylation levels assessment as first-line molecular assay, as preliminary results showed that differential methylation profiles could be suggestive of the presence of a reduced D4Z4 allele, genetic mutations in disease modifier genes or in genes related to other forms of myopathies.
The improvement of FSHD molecular diagnosis would be highly beneficial to patients and families, which could undertake diagnosis-specific interventions, participate to clinical trials and be able to make informed reproductive choices. Moreover, the comparison of genetic results with clinical and instrumental data may be helpful to perform accurate genotype-phenotype correlations, which may support physicians in the diagnosis, prognosis and selection of the possible therapeutic treatments or physiotherapy approach. In addition, the enhancement of diagnostic procedure and power may also contribute to the advancement of translational research activities tailored to increase the knowledge of FSHD pathophysiology and progression and to develop novel treatment strategies.
FSHD is muscular dystrophy affecting roughly 1/8000 individuals with a complex clinical profile and poor understanding of the molecular mechanisms causing the disease. For these reasons, until now, no efficient pharmacological treatments are available. The majority of current therapies aim to enhance patient’s quality of life reducing the disease progression. The discovery of DUX4 as a driving factor in FSHD opens new perspectives in the development of specific therapeutic strategies. Several approaches have been considered to reduce DUX4 toxicity including the genetic regulation of DUX4 expression by genome editing methods, the inactivation of DUX4 mRNA by siRNA or antisense oligonucleotide, and the interference of DUX4 protein activity by DNA aptamers. In this context, the use of mimicking peptides (mPeps) able to modulate the activity of DUX4 could represent an appealing DUX4 modulation strategy, whose feasibility and potentiality are largely underexplored. mPeps are synthetic molecules that specifically recognize their interacting partners and that can be easily produced and delivered in a specific cellular compartment. For these features, they are considered promising candidates for the modulation of PPI associated to human disease and for the development of novel therapeutic interventions. Thus, the identification of MATR3 as an endogenous inhibitor of DUX4 transcriptional activity offers a new valuable opportunity in FSHD therapy that relies on the design of mPeps selectively targeting the DUX4 toxic activity. Results from this project will offer the possibility to generate new drug-like molecules for the treatment of FSHD.
FSHD is the third most prevalent human muscular dystrophy, and there are no accepted treatments. FSHD muscle is characterized by the progressive accumulation of fat and fibrotic tissue, which severely compromises muscle physiology.
We will investigate whether the expression of DUX4 changes the exosomes profiling of the muscle fibers, and whether this change facilitates the apposition of adipocytes and fibroblasts in the muscle. We will identify the RNA content of the muscle-released exosomes in an FSHD mouse model combined with an exosome-tracer system in vivo.
Results of this study will represent the basis to design new potential treatments to slow down FSHD progression.
The proposed work will investigate an early and central step of DUX4 toxicity - the direct protein interactions through which it mediates gene regulation. Our preliminary data shows that our current understanding of this step is partial. Therefore, the significance is twofold:
1) Obtaining a more complete picture of the FSHD pathology.
2) Providing new intervention points through which the toxicity may be modulated.
Progressive muscle wasting significantly reduces quality of life in FSHD patients. DUX4 expression and the initiation of apoptosis are prevalent in FSHD muscle, but it is currently unclear if apoptosis occurs in fibres themselves, or if the apoptotic signal is contributed by other cell types residing in the tissue. However, FSHD muscle fibres show clear signs of atrophy (reduced cross-section diameter).
Understanding the atrophy-related pathology of FSHD muscle will likely provide new therapeutic entry points to treat the disease or slow the progression. Currently, experimental therapies are focused on reducing DUX4 expression, or inhibiting downstream targets inducing apoptosis. By elucidating the pathway downstream of DUX4 expression and understanding atrophy in FSHD skeletal muscle fibres as well as the influence of satellite cells on the pathology, we aim to identify novel therapeutic targets. Additionally, this work has the potential to facilitate the repurposing of drugs/exercise regimes that are used to combat muscle atrophy in sarcopenia (10.4235/agmr.19.0028) or cachexia (https://doi.org/10.1002/rco2.30).
Facioscapulohumeral muscular dystrophy (FSHD) is a common neuromuscular disease triggered by misexpression of DUX4 that leads to poorly understood multi-cellular responses that contribute to muscle degeneration. It has been seen as a limitation that current FSHD mouse models are based on the human gene expression (DUX4) in the murine organism. Thus, as a necessary addition to current models, we generated a novel transgenic mouse inducible for mouse Dux, a mouse ortholog of DUX4, in skeletal muscle for studying FSHD pathology in vivo at molecular and cellular levels.
We recently demonstrated that DUX4 is a corepressor of the progesterone and glucocorticoid nuclear receptors. Thus, in addition to its role as an activating transcription factor, DUX4 could contribute to downregulate gene expression via co-repression of hormone nuclear receptors. The study of this hypothetical endocrine role is relevant to the knowledge of the normal role of DUX4. It could also unveil the potential connection between pathological expression of DUX4 in muscle and the gender differences and muscle inflammation observed in FSHD. These studies may have relevance in the current rational approaches for the treatment of FSHD patients.
FSHD is a muscle disease caused by aberrant overproduction of the protein DUX4. The mechanism through which DUX4 causes the disease is incompletely known. Recently, the inflammatory molecule IL-6 was identified as the first marker significantly associated with FSHD disease duration, severity, and muscle weakness. While the current data suggest that excessive IL-6 levels could contribute to FSHD and could be targeted for therapeutic purposes, the relevance of IL-6 for the disease has not been directly tested and the source of its production is unknown. Recent data also suggest that alteration in fibro-adipogenic progenitor (FAP) cells could contribute to the disease, but the mechanism responsible for FAP alteration in FSHD is unknown. My preliminary data provide a possible molecular explanation for the above changes. In this project, I propose a comprehensive analysis combining various approaches to identify the sources of IL-6 production and evaluate the therapeutic relevance of blocking the pathway for the treatment of FSHD.
FSHD is less prevalent in the pediatric and adolescent population however its impact on the social, emotional and physical functioning of affected children and adolescents is significant. Research, outcome measure development and the establishment of biomarkers in preparation for clinical trials is well advanced in adults with little research in children and adolescents. Preparation for clinical trials in this younger FSHD cohort is hampered by little published data outlining the natural history of disease progression, poor adherence to standards of clinical care and the paucity of reliable, valid and responsive disease specific measures of function.
The availability of longitudinal data documenting disease natural history will advance clinical trial readiness in a younger FSHD cohort. Comparison of these data with already recognized care recommendations for adults will enable researchers to establish whether the development of pediatric specific standards of care are required. Clinical trial design has been hampered by the paucity of measurement property evidence to support outcome measure responsiveness when evaluating individuals with FSHD. The collection of serial MRI, physical function and quality of life data using already validated and novel outcome measures, will enable researchers to estimate their responsiveness when evaluating disease progression. The neurophysiological profiling proposed in this study and the investigation of any links to physical, cognitive and social function will increase our understanding of disease and contribute to holistic clinical care of young people with FSHD.
This study will contribute much needed data to support and advance our clinical care and clinical trial readiness in children and adolescents with FSHD.
2021 awards
The FSHD Society initiated a circulating biomarker study aimed at measuring DUX4-regulated target gene quantification in patient blood using existing bioassays. Several experiments have been designed to measure protein products of DUX4-regulated genes in cell culture experiments and to confirm if their levels are modulated by DUX4 expression levels. If successful, FSHD patient and unaffected volunteer blood samples will be tested to see if differences between the groups are detectable and whether a blood test could substitute for more common needle biopsies. These studies will be carried out in collaboration with Drs Amy Campbell and Sujatha Jagannathan at the University of Colorado Anschutz Medical Campus, Denver, CO; patient and unaffected volunteer patient blood samples will be provided by Dr. Rabi Tawil at the University of Rochester Medical Center and quantitative proteomic services will be provided by Olink, a proteomic assay service provider. The total estimates for these studies is $32,892.
The objective of this study is to evaluate the prevalence of swallowing difficulties, bowel symptoms, and urinary symptoms in people with FSHD. Methods: We are using an anonymous online survey sent to the FSHD Society mailing list, to ask patients and household controls about swallowing difficulties, bowel symptoms, and urinary symptoms, including their frequency, severity, and impact on quality of life, in addition to what (if any) medications were needed to treat those symptoms. There have so far been 740 participants who completed the questionnaire. Outcomes: We will use the collected data to determine if the symptoms of interest are in fact more prevalent in people with FSHD and, if so, to what degree those symptoms impacted on quality of life. These results may help guide us in determining if further investigation is necessary regarding causes and/or treatments of these symptoms in people with FSHD.
The FSHD Society is providing $24,310 in support to the University of Rochester Medical Center for the purchase of an ultra-low temperature freezer and access to a Laboratory Information Management System (LIMS) to support and maintain the expansion of the FSHD Clinical Trail Research Network (CTRN) tissue biobank. The biobank will be a critical asset for the development of exploratory biomarker studies, as well as any future related validation work.
One of the most frequent symptoms of FSHD is weakness of the facial muscles. Weakness of the facial muscles limits facial expression and many patients report it as a major disabling symptom, because it hinders their non-verbal communication. Despite the frequency and relevance of facial weakness in FSHD, thorough knowledge on its progression and consequences is lacking. This is mostly due to the fact that until recently, there were no adequate measurement tools to measure facial weakness. In this study, we use four different newly developed measurement instruments to evaluate how facial weakness changes over time. These instruments include a score for the degree of weakness applied by a physician, an automated video-based system to grade the degree of weakness, ultrasound to assess the muscle tissue, and a patient-reported questionnaire on functional consequences. Approximately 100 FSHD patients are followed for up to five years. The longitudinal data collected in this study will shed light on the pattern of involvement of the facial muscles, progression over time, and how weakness relates to the symptom burden. This knowledge can be used to improve patient counseling in the clinic, as well as enable the development and testing of therapies for facial weakness.
DUX4 belongs to a class of genes known as transcription factors. These genes act like switches that tell the cell when other genes need to be turned on or off. DUX4 normally functions in switching on genes during early embryonic development, but when it is turned on in adult muscle it becomes toxic, leading muscle fibers to die over time. While we know the identities of the genes that DUX4 switches on, we do not know which of them cause toxicity. This information is critical, as it would allow us to design new molecular therapeutics. Here, we will investigate which genes cause toxicity by comparing the genes activated by DUX4 with genes activated by other relevant transcription factors. For example, other DUX-family genes are similar to DUX4, and likely activate similar gene sets, but they are not toxic. By comparing the gene sets that they activate with DUX4’s, we can make a list of potentially toxic genes. We will identify the gene sets activated by these and other factors and use them to find the toxic genes DUX4 activates. We will then design molecular tools to silence the toxic genes, and evaluate their potential for the clinic.
A number of laboratories in academia and biopharma are now developing treatments for FSHD that have a significant chance of succeeding in slowing or even stopping the progression of the disease. The question is how to evaluate each therapy accurately, to pick the one or two that are most promising for further study. The best way of doing this is with a “biomarker”, typically a molecule that increases in amount when the disease is fulminant and decreases in amount when the disease is quiescent or under effective treatment. Ideally, a biomarker molecule should be accessible in the serum or the urine, so that patients are not discomforted when tests for it are needed. We have found that one of the proteins that increase in amount in FSHD as a result of the DUX4 program, called SLC34A2, has many of the properties of a biomarker for FSHD. Here we propose to evaluate its potential usefulness as a biomarker, with the hope that it will eventually be of value in clinical trials. The methods we establish should be applicable to other potential biomarkers as well.
In a therapeutic approach for FSHD, we have developed antisense oligonucleotides (ASOs) that bind to DUX4 messenger RNA and prevent production of the toxic DUX4 protein. However, the use of ASOs to treat myopathies is currently limited because after injection they are rapidly cleared from the bloodstream by the liver and kidneys and if they reach other tissues they do not preferentially enter into muscles. Our general aim is to protect these ASOs into nanoparticles and link these to a moiety that will direct ASO delivery to muscle cells after injection into the bloodstream. We have previously identified 5 short protein fragments (peptides) that present a preferential entry into muscle as compared to non-muscle cells, thus putative muscle-specific peptides (MSPep). We want here to determine whether hooking an MSPep to an ASO could improve its transportation in the blood and its muscle uptake, leading to an efficient reduction of toxic DUX4 protein. Our project will determine in cell culture and mouse models #1 the muscle uptake, cell entry pathways, and body distribution of MSPep-ASO; #2 whether the ASO can be released from MSPep-ASO inside the muscle cells; #3 whether it can efficiently reduce toxic DUX4 expression in muscles.
Facioscapulohumeral muscular dystrophy (FSHD) is caused by mutations that turn on the toxic DUX4 gene in skeletal muscle where it causes muscle degeneration. The cellular mechanisms that turn on DUX4 are poorly understood. A more detailed description of how DUX4 gene is turned on is essential to identify drug targets for suppressing DUX4 in FSHD. The discoveries by Dr. Fran Sverdrup’s group and Fulcrum Therapeutics showed that p38 MAP kinase is a key driver of DUX4 expression not only reveal a therapeutic opportunity (p38 inhibitor losmapimod). Inhibition of p38-MAPK effectively suppresses myotoxic DUX expression in cellular and animal models of FSHD, paving the way for the clinical evaluation of the p38 inhibitor losmapimod in FSHD patients. We propose to exploit this finding to identify regulatory factors that turn on DUX4 and determine how p38 dependent and independent signaling links to DUX4 expression during muscle differentiation.
Our group studies a protein called SMCHD1, which is able to keep genes in a ‘sleeping’, or silenced, state. It has been shown that FSHD patients can have variations in the SMCHD1 gene, causing it to misfunction. This leads to an inappropriate ‘awakening’, or activation, of a toxic DUX4 gene, leading to muscle cell death and the associated muscle wasting and weakness that is experienced by FSHD patients. In this project, I will investigate the currently unknown three-dimensional structure and function of the SMCHD1 protein. By doing so, we can reveal the mechanistic details that allow SMCHD1 to perform its regular function, and inform us on how FSHD-associated variations in the protein can directly alter its activity. First, I will employ cutting-edge microscopy techniques that allow the direct visualisation of the SMCHD1 protein. Secondly, I will use real-time imaging methods to monitor and compare the behaviour of healthy versus FSHD-affected SMCHD1 proteins. We anticipate that by uncovering the FSHD-associated changes in SMCHD1’s function we can learn how to manipulate the protein’s activity to make it more efficient at silencing the toxic DUX4 gene that causes FSHD.
When healthy muscle cells grown in the laboratory are forced to make the FSHD-causing protein DUX4, they die catastrophically. Early on during DUX4 expression, an essential quality control pathway in the cell that keeps defective proteins from being produced from aberrant messenger RNA molecules fails. As a result, two things happen. One, DUX4 mRNA, which is a natural target of this quality control pathway, becomes amplified. Two, other aberrant mRNAs in the cell become stabilized, flooding the cells with toxic proteins. Both of these consequences can play significant roles in FSHD pathogenesis. In this proposal, we seek to determine the mechanism by which the RNA quality control pathway fails upon DUX4 expression with the goal of rescuing its activity as a therapeutic intervention for FSHD.
FSHD patients suffer from progressive muscle weakness and wasting. Although the cause is production of the toxic protein DUX4, it is unclear how DUX4 causes muscle disease. Recent studies show that FSHD patients have impaired metabolism, a condition called metabolic stress. Mitochondria are energy factories inside cells, and are crucial to supply energy for muscle to work. When mitochondria fail to function efficiently, muscle develops a state of energy crisis and degenerates. We found that DUX4 changes the way mitochondria work, so that they start to produce radicals, small but highly reactive molecules. Radicals interfere with muscle metabolism and lead to muscle impairment, especially when they disturb how muscle uses oxygen. This changes how FSHD muscle reacts to low oxygen availability (e.g. as occurs naturally during exercise). Our preliminary studies show that antioxidants that specifically target radicals produced by mitochondria can restore FSHD muscle cell function. We will test food supplements/drugs that act on specific radical-controlled cellular systems to find new therapies that can alleviate FSHD. Thus, our investigation of FSHD muscle metabolism will both broaden our understanding of how DUX4 causes FSHD, and identify new therapeutics to complement more experimental therapies directed at reducing DUX4 levels.
While aberrant expression of the transcription factor DUX4 has been proposed to cause FSHD, the factors that DUX4 expression in FSHD skeletal muscle remain poorly understood. Previous studies have shown that cellular stress can stimulate DUX4 expression and contribute to onset or progression of FSHD muscle pathology. Muscle membrane injury is a repetitive and routine form of cell stress that develops as a result of muscle contraction. Rapid repair of this membrane injury is essential for muscle health, and deficits in membrane repair contribute to the progression of other common muscular dystrophies. We have recently found membrane repair deficits in FSHD human muscle cells, and in muscle fibers from a transgenic mouse model of FSHD. Additionally, we have found preliminary evidence that cell membrane injury can trigger DUX4 expression. In this proposal, we aim to examine how cell membrane injury promotes changes the organization and behavior of the regulatory elements that influence gene expression in response to injury in FSHD and healthy cells. This study will help elucidate the potential role of plasma membrane injury in the pathophysiology of FSHD in skeletal muscle, and may identify novel targets for therapy to improve membrane repair or mitigate DUX4 expression.
Lay Summary: There are currently limited therapeutic options for FSHD, in large part due to lack of understanding of disease mechanism and druggable molecular targets. We recently discovered that a specific metabolic response triggered in the muscle cells of FSHD patients ultimate contributes to their death. We would now like to perform further studies to assess whether these metabolic changes can be predictive of FSHD pathology in various cell models and animal tissues, and to test existing drug compounds targeting this response that can act as a new class of compounds for the treatment of FSHD patients. (Note: Angela Lek has left Yale in fall of 2021; the PI on this project is now Monkol Lek, PhD.)
FSHD is caused when the DUX4 gene turns on in muscle. The DUX4 gene product is toxic to muscle cells, and over time, enough damage can accumulate to cause muscle weakness associated with FSHD. Therefore, most FSHD therapy studies are now focused on turning the DUX4 gene “off” in FSHD muscle. We developed a system that can attack the DUX4 messenger RNA and destroy it before it is used to produce the toxic DUX4 protein. The system we used relies upon the Cas13 enzyme, which can be directed to cut RNA molecules in cells. We found that the Cas13 system could inhibit DUX4 expression by more than 85% in FSHD patient-derived muscle cells. This system was also able to reduce DUX4 expression in an FSHD mouse model by 50%. In this study, we are trying to improve the Cas13 system to allow greater DUX4 silencing in mouse muscle, and ensure the safety of the approach prior to translating it to humans.
The DUX4 gene is normally repressed in skeletal muscle and its abnormal expression leads to FSHD. Mouse models of human disease are useful for answering questions about disease pathology and for identifying therapeutic avenues. However, mice do not have the DUX4 gene so FSHD model mice need to be engineered by inserting the DUX4 gene into their genome. Mice that contain the DUX4 gene already exist, however, that gene is not under its normal regulation found in humans. In this study, we will generate novel FSHD model mice using the entire FSHD region of human chromosome 4q35, including the DUX4 gene and its neighboring genes as well as its own regulatory elements, all derived from an FSHD patient who has DUX4 expression and pathological phenotype in skeletal muscle. We will investigate whether DUX4 is abnormally expressed in skeletal muscle and if its expression could lead to the myopathic phenotype mimicking what is seen in the FSHD patients. Successful completion of this study will provide insight into the mechanisms of DUX4 expression in skeletal muscle of FSHD patients, and this animal model will be used for developing and validating FSHD therapeutics for academia and industry.
The Facioscapulohumeral Muscular Dystrophy Clinical Trial Research Network (FSHD CTRN) is a consortium academic research centers in both the United States and Europe with expertise in FSHD clinical research, or in conducting neuromuscular clinical trials. The FSHD CTRN helps close gaps in trial readiness, and also provides a network of sites with a centralized streamlined regulatory process, specific, common expertise in FSHD, and an engaged patient population ready to conduct efficient, high quality clinical trials. Because of its prominent role in clinical trial readiness for FSHD, additional funding is being provided to expand the existing consortium in the United States with four additional sites (U. Florida; U. Texas Southwestern Medical Center; U. Colorado, Stanford U. School of Medicine). This funding aims to ensure better coverage and access to patients, as well provide additional resources for the effective management and coordination across the entire network.
2020 grant awards
The Facioscapulohumeral Muscular Dystrophy Clinical Trial Research Network (FSHD CTRN) is a consortium academic research centers in both the United States and Europe with expertise in FSHD clinical research, or in conducting neuromuscular clinical trials. The FSHD CTRN helps close gaps in trial readiness, and also provides a network of sites with a centralized streamlined regulatory process, specific, common expertise in FSHD, and an engaged patient population ready to conduct efficient, high quality clinical trials. Because of its prominent role in clinical trial readiness for FSHD, additional funding is being provided to expand the existing consortium in the United States with four additional sites (U. Florida; U. Texas Southwestern Medical Center; U. Colorado, Stanford U. School of Medicine). This funding aims to ensure better coverage and access to patients, as well provide additional resources for the effective management and coordination across the entire network.
FSHD is one of the most common muscular dystrophies and yet little is known about the risk of various functional motor outcomes, like the risk of ending up in a wheelchair, or the risk of having to use non-invasive ventilation. This study seeks to determine the risk of functional motor outcomes, and those characteristics present at baseline (like age, gender, mutation, or baseline functional status) which may predict a future change in function. We propose to use two techniques – one where we look at the incidence of predefined outcomes using expert medical knowledge to inform our analysis; and a second technique where we use all the data in the registry, and modern machine learning algorithms to determine which information can predict specific functional motor outcomes, like requiring a wheelchair for ambulation. We will compare the results of both approaches, and together they will help inform not only our clinical care of patients, but also inform what information is most important to collect for future registries and help us plan for therapeutic interventions.
The FSHD field requires a reliable protein biomarker for FSHD to facilitate diagnostic and therapeutic testing. Our laboratory has reported that the protein encoded by SLC34A2, a gene upregulated by DUX4, is present in biopsies of FSHD muscle at much higher levels than it is in biopsies of healthy muscle (Mueller et al., 2019). Control human serum does not contain detectable levels of the protein. Here we propose preliminary studies to compare two commercial antibodies to SLC34A2, along with one antibody we already used extensively to pilot the development of a sensitive “sandwich” ELISA assay. This would lay the groundwork for rapid and quantitative testing of the protein in biological samples with the goal of using it in the clinic.
miRecule Inc. is a company specializing in targeted delivery of chemically modified RNA therapeutics. These types of therapies have proven to be effective at treating rare muscle diseases such as Duchenne muscular dystrophy. However, a significant hurdle for nucleic acid therapeutics is designing an efficient platform that can deliver an effective dose of these large molecules across the membrane of target cells. The pathogenesis of facioscapulohumeral muscular dystrophy (FSHD) is primarily caused by the aberrant expression of a normally dormant gene, DUX4. Several studies have shown that RNA therapeutics can be effective at knocking down DUX4 expression in muscle tissue and alleviating the pathology of FSHD in animal models. We have a platform whereby modified human antibodies are conjugated to RNA therapeutics to target delivery to specific tissues, including skeletal muscle. We propose to develop, apply and validate our platform technology to developing a treatment for FSHD.
Genetic testing to identify the D4Z4 contraction responsible for FSHD is difficult due to the large size of the array and the nearly identical sequence in another chromosome. The gold-standard diagnostic test is Southern blot after restriction digest which differentiates the chromosome 4q and 10q D4Z4 arrays and estimates their length. More recently, molecular combing and optical mapping techniques have emerged. These methods represent a significant advance, but like Southern blotting are technically demanding and are only available at a few select laboratories and neither method provides information about the methylation state of the locus. We propose to optimize a recently developed DNA fragment enrichment tool as a comprehensive diagnostic tool for FSHD that will determine the length of the 4q D4Z4 array, differentiate the 4q from 10q array, determine A/B haplotype, and estimate methylation across the locus.
The U.S. National Registry for Facioscapulohumeral Muscular Dystrophy (FSHD), is the largest and longest running national FSHD patient registry with up to 19 years (Average, 6 years) of prospective follow up information on 950 individuals with FSHD with an average of about 60 additional patients joining the registry every year. The Registry has been used extensively to study various aspects of FSHD including pain, incidence of retinal vascular disease, overall disease progression, genotype phenotype correlation, development of FSHD-specific patient reported outcome measures, natural history, recruitment for and an infantile onset FSHD study, and recruitment of patients for translational studies at UR, KUMC and UW. Most recently, the FSHD registry data was examined using AI to look for correlations between disease severity and a number of other variables.
Since the initiation of the Registry several initiatives were started to improve data quality but also increase the number of FSHD patients who are genetically confirmed as testing was expensive and often not covered by insurance. Genetic confirmation has become more critical now that target treatment trials are underway and genetic confirmation is one of the entry criteria. With funding from the FSHD Society and Friends of FSH Research we have been able to genetically confirm FSHD genetically in an additional 90 Registry participants.
Another initiative currently underway to facilitate participation in the Registry is to fully digitize the Registry enabling participants to fill out the forms online directly rather than via paper forms. This improves efficiency cutting on the cost of mailing and making the transaction with Registry participant easier. It also improves efficiency on the back end lessening the need for manual data entry from paper forms to the digital database. Currently, the Registry uses a REDCap data base. The process of creating a patient Red Cap interface is underway but is hampered by the lack of specific funding for this task. Work has therefore been slow and intermittent as other funded projects receive priority.
Most studies on FSHD are concentrated on muscle cells only. In muscles, myotubes and satellite cells exist in a contact with a variety of other cells and this microenvironment plays an important role in muscle homeostasis. The muscles of FSHD patients are subjected to inflammation, invasion with non-muscle cells and fibrosis. We will study the role of this environment on muscle differentiation and homeostasis in the FSHD context. The answers that we will provide during this study will help to improve our knowledge of the pathophysiology of FSHD and envisage new potential treatment approaches.
2019 grant awards
FSHD is a complicated disease, and it has taken many decades of research to understand the underlying cause. Fortunately, we now know that FSHD is caused by switching "on" the DUX4 gene in muscle. This is an abnormal occurrence, as DUX4 is supposed to be "off", and when DUX4 is turned on, it damages muscle cells. Today, there are several new promising therapies emerging that are designed to shut off DUX4. All of these need to be tested in people to make sure they are effective. For therapies that target DUX4, one way to know they work is to measure DUX4 levels in FSHD muscles. Unfortunately, this is not currently possible, as DUX4 is hard to detect and there are not methods available to easily find DUX4 in muscle. Our project is designed to test a new DUX4 detection method. We already showed this works in cells on a dish, and now we need to demonstrate that we can find DUX4 in human muscles donated by FSHD patients. This is the major goal of the project. If successful, we will then begin to apply this method so it is useful for testing DUX4 levels in FSHD therapy trials.
Facioscapulohumeral muscular dystrophy (FSHD) is the most prevalent progressive myopathy that afflicts both children and adults regardless of the gender. FSHD is caused by aberrant gain of expression of the double homeobox 4 (DUX4) gene causing toxic effects in muscle cells. Despite the consensus on the pivotal role of DUX4 and several clinical trials, there is currently no cure or an effective therapeutic approach for FSHD patients. In our studies we identified a novel regulator of DUX4. Targeting this factor allows to block DUX4 expression and rescue the behavior of muscle cells from FSHD patients. Our proposed studies will investigate the idea of a novel approach that could represent a promising therapeutic option for FSHD patients.
While aberrant expression of the transcription factor DUX4 has been proposed to cause FSHD, very little is known about what factors trigger DUX4 expression in FSHD skeletal muscle. Previous studies have shown that cellular stress can stimulate DUX4 expression, suggesting stimuli that augment cell stress could play a central role in the onset or progression of FSHD muscle pathology. Muscle membrane injury is a repetitive and routine form of cell stress that develops as a result of muscle contraction. Rapid repair of this membrane injury is essential for muscle health, and deficits in muscle membrane repair contribute to the progression of other common muscular dystrophies. We have recently found similar membrane repair deficits in FSHD human muscle cells, and in muscle fibers from a transgenic mouse model of FSHD. Therefore, this proposal will test the hypothesis that that molecular stress caused by plasma membrane injury exacerbates FSHD by inducing DUX4 expression, and by altering gene expression and gene regulation in FSHD muscle cells. This study will help elucidate the potential role of plasma membrane injury in the pathophysiology of FSHD in skeletal muscle and may identify novel targets for therapy to improve membrane repair or mitigate DUX4 expression.
Symptoms that FSHD patients first notice can be very different, as can the sequence of muscles affected. Identical twins frequently report differences, with one twin very affected while the other is not. Understanding what causes these differences is key to identifying lifestyle changes and special treatments that can minimise FSHD severity.
To understand such differences, we need to categorise FSHD patients into groups depending on the order that symptoms appear and compare them to identify modifiable factors protecting some patients. We performed a categorisation using the UK FSHD registry, evaluating 222 patients, identifying 4 types of FSHD with respect to muscle groups first affected (face, shoulder or leg/foot). This project is a global collaboration to expand these findings, integrating large patient databases from Italy and USA to establish the largest FSHD patient dataset. We will analyse this dataset to identify FSHD patient subtypes and modifiable factors contributing to milder disease, using state-of-the-art computational techniques. This will give an accurate description of how FSHD progresses in an individual, allowing better planning for future needs, and highlight beneficial lifestyle changes. In the mid-term, molecular research into the patient subtypes will improve understanding of what causes FSHD and inform individualised approaches to therapy.
Harnessing CRISPR technology, Lek found molecular pathways that rescued FSHD muscle cells (in a test tube) from the deadly effects of the DUX4 gene. She identified drugs that mimic this protective effect and plans to test them further in zebrafish and mouse models of FSHD. “Our aim will be to identify and prioritize testing of compounds with minimal side effects, are suitable for long-term dosing and have FDA approval [for other conditions] to ensure a fast route to a clinical trial in patients,” she said.
The ubiquitin-proteasome system (UPS) is responsible for degrading 80 to 90 percent of proteins in cells, and has been shown to play an important role in mediating muscle atrophy. DUX4 expression in muscle precursor cells changes the expression of many genes involved in the UPS. Homma proposes to identify the possible mechanisms by which DUX4 may promote muscle atrophy. Results of this project might reveal therapeutic targets for DUX4-induced muscle atrophy.
The Jones lab recently identified the epigenetic regulator ASH1L as a key driver of pathogenic DUX4 expression, and showed that reducing ASH1L levels nearly abolishes DUX4 expression without significantly altering expression of other genes. “We propose that ASH1L is an outstanding FSHD therapeutic target,” said Jones. The lab intends to identify promising lead inhibitors of ASH1L activity.
Female FSHD patients are clinically less affected than males and present a higher proportion of asymptomatic carriers. Progesterone is a key female hormone involved in regulating the uterine lining. The lab found that DUX4 is a co-repressor of the progesterone nuclear receptor (NR) in various cell models. These results suggested that DUX4 could indirectly modulate gene expression by repressing the activity of the progesterone NRs, a previously unrecognized role for DUX4.
Bridge funding for CTRN Project Manager in the department of Neurology at the University of Kansas Medical Center. The Project Manager will be responsible for coordinating overall network wide activities and research studies. Duties will include creating personnel rosters for each committee, helping organize and create CTRN governance documents and SOPs, common data elements, interacting with site coordinators, data personnel, regulatory specialists, maintaining CTRN website, managing active studies and new study inquiries, and helping coordinate projects at the network level. In addition, Project Manager will work with KUMC and advocacy-based patient engagement on recruitment, and retention efforts, engaging underserved populations, as well as working with the data office to ensure data quality across the CTRN network studies. For the network expansion to additional US sites, Project Manager will ensure the availability of local resources in expansion sites, ensure each site adheres to training, and meets CTRN goals.
2018 grant awards
Project Summary. Facioscapulohumeral muscular dystrophy (FSHD) is a progressive neuromuscular disease that diminishes the quality of life for hundreds of thousands of people throughout the world. Current evidence indicates that FSHD is caused by mis-expression of the DUX4 transcription factor in skeletal muscle fibers, which leads to skeletal muscle cell death and weakness. One important consequence of skeletal muscle DUX4 expression is the downregulation of a conserved RNA quality control pathway, called nonsense mediated RNA decay (NMD). Compromised NMD in FSHD skeletal muscle cells results in increased levels of aberrant mRNAs that contain premature translation termination codons (PTCs) and endogenous mRNAs that encode stress-inducible, pro-apoptotic factors. In the studies proposed here, we will 1) determine whether PTC-containing mRNAs are translated to potentially deleterious truncated proteins, and 2) whether rescuing NMD function is capable of preventing cell death by suppressing levels of pro-apoptotic factors. Thus, the studies proposed here will further elucidate the mechanisms by which reduced RNA quality control contributes to skeletal muscle deterioration in FSHD. Importantly, these studies may inform the development of novel biomarkers or pharmacologic therapies in FSHD.
Significance: A project proposed by a young promising post-doctoral fellow focused on providing insight into the molecular underpinnings of FSHD, which may lead to the development of novel diagnostic biomarkers and therapeutic strategies. Done by determining the consequences of diminished RNA quality control in FSHD skeletal muscle. Previous studies by this lab have found that DUX4 expression in skeletal muscle leads to severe perturbation of an evolutionarily conserved RNA quality control pathway: nonsense mediated decay (NMD). In healthy skeletal muscle, NMD plays a beneficial role in surveying and eliminating aberrant RNA molecules, as well as suppressing levels of stress response proteins that can cause cell death. Their preliminary data indicate that perturbed NMD in FSHD skeletal muscle leads to increased levels of aberrant RNAs, hyperactivation of cell stress response pathways, and muscle cell death. These findings reveal that diminished RNA quality control is a pivotal event that contributes to skeletal muscle deterioration in FSHD. Project might help clarify if rescuing NMD function can slow or prevent skeletal muscle deterioration in FSHD.
Antisense oligonucleotide (AON) therapy shows promise for treating an array of disorders, however, several issues associated with AONs affect its applications, including 1) difficult in systemic drug delivery because these AONs could not easily cross the lipid bilayer of cells; 2) harmful off-target effects and toxicities; 3) low stability due to degradation by intracellular and extracellular nucleases.; and 4) immune responses via toll-like receptors. The 2’-O-methoxyethyl (2’MOE) and locked nucleic acids (LNAs) modification are two widely used chemistries for designing gapmer-type antisense oligonucleotides, which overcome many of the issues associated with AONs. LNAs have the 2’,4’-methylene bridge and 2’MOEs have a simple methoxyethyl substituent attached to the 2’ oxygen. These modifications have been shown to enhance target binding affinity, specificity, and resistance to degradation by nucleases. The LNA gapmers provide a stronger affinity in comparison to many other modifications; therefore it is possible to design shorter gapmers for the same efficacy. This design will also increase the uptake by gymnosis in the absence of any carriers or conjugation. Compared to LNA gapmers, 2’MOE gapmers may be less potent; however, 2’MOE gapmers are likely to be safer than LNA gapmers due to less off target effect (the 2’MOE gapmers are slightly longer than the LNA gapmers). Considering the pros and cons, we propose to examine and compare the LNA and 2’MOE gapmers that target the same region of the DUX4. We have designed three 2’MOE gapmers and conducted in vitro studies. We showed superior knockdown efficiency in vitro when tested in immortalized FSHD myoblasts. Given the properties mentioned above and their proven track record in the clinic, it would be favourable to design 2’MOE gapmers for FSHD treatment if the 2’MOE gapmers have a similar potency in vivo and less toxicity. In the proposed studies, we will study the efficacy and safety of the 2’MOE gapmers in vivo and compare to the LNA gapmers. We are currently characterizing the LNA gapmers using both in vitro and in vivo model, which is supported by the FSH society. In this proposal, we would like to request support for purchasing 2’MOE gapmers and conduct in vivo efficacy studies, the data will be compared with the data from our LNA studies. The findings will allow us identify the lead compound for treatment development for FSHD. In the Aim 1, we will systemically delivery the 2’MOE gapmers to the FLExDUX4 mice and determine the efficacy of the treatments. In Aim 2, we will evaluate off-targets, immunogenicity and toxicities and compare the data to the LNA gapmer data. The proposed studies will allow prioritizing and identify the most promising gapmers for drug development.
Significance: Antisense oligonucleotide (AON) therapy shows promise for treating FSHD, however, several issues arise with AONs including 1) difficulty in systemic drug delivery; 2) harmful off-target effects and toxicities; 3) low stability due to degradation; and 4) immune responses. The 2’-O-methoxyethyl (2’MOE) and locked nucleic acids (LNAs) modification are two chemistries for designing gapmer-type antisense oligonucleotides, which overcome issues associated with AONs. Previous support by the FSH Society helped this lab in developing an effective antisense oligonucleotide (AON) strategy to target DUX4 and reduce its expression. The LNA gapmer under investigation was able to effectively knockdown DUX4 both in cell culture and mice. In the FSHD mouse model generated by Dr. Peter Jones’ group, Chen showed functional recovery of muscle strength after systemic delivery of the LNA gapmers. In this proposal, they will compare the LNA gapmers to 2’MOE gapmers which are targeting the same target sequences of the DUX4. The 2’MOE was recently approved for treating spinal muscular atrophy by the FDA. It is considered less potent but safer than LNA. In an in vitro experiment done by collaborator, Dr Yokota, he showed that the 2’MOE gapmers targeting the same DUX4 region effectively knocked down the DUX4 transcripts. In this proposal Chen will compare the in vivo efficacy and the safety of the 2’MOE gapmers to the LNA gapmers. The goal is to carefully characterize and identify the compound that will be moved forward for drug development.
FSHD presentation is non-uniform, and there may be extreme variability in severity of symptoms, rate of progression and age at onset, even in families with several affected relatives. Similarly, asymmetrical weakness is common. It has been hypothesized that this non-uniformity of presentation might be due to the regulation of DUX4 expression by yet undetermined factors. Although some of the genes that modify DUX4 gene expression are already known (e.g., SMCHD1, DNMT3B), overall the regulation of DUX4 gene expression is still relatively unclear, and genes that directly target DUX4 mRNA have not been identified. We think that DUX4 gene expression modifiers might influence DUX4 toxicity and FSHD disease penetrance. In previous proposals to the FSH Society, our central hypothesis was that some endogenous microRNAs (miRNAs) could target the DUX4 transcript, thereby reducing DUX4 expression and toxicity. During the past two years, we have been investigating this hypothesis. In particular, we have been investigating the action of a long non-coding RNA (H19) and its miRNA by-product (miR-675) against DUX4. So far, my recent investigation provides the first proof for H19 and miR-675 reducing DUX4 expression and toxicity, which paves the way to develop new therapeutic approaches by targeting or using natural miRNAs such as miR-675. More specifically, my aim here is to expand our pipeline of DUX4-targeted miRNA-based gene therapy for FSHD by using miR-675 as a new miRNA-based gene therapy candidate. This proof-of-principle also supports the identification of the full set of natural DUX4-targeted miRNAs that would represent a set of potential miRNA therapeutics or drug targets. Our project has two aims. The first aim focuses on identifying the full set of natural miRNA that could target DUX4. In this aim, we would ideally like to tie one or multiple natural miRNAs into FSHD disease progression, but even if we are unable to find evidence for miRNAs acting as DUX4 modifiers, we propose they could still be used as potential therapeutics. The second aim focuses on performing a pilot study to develop a DUX4-targeted miR-675-based gene therapy for FSHD. Specific Aim 1: To functionally identify every natural human miRNA capable of targeting DUX4 in vitro. Specific Aim 2: To develop a DUX4-targeted miR-675-based gene therapy for FSHD.
Significance: Dr. Saad has with FSH Society funding for past two years been investigating endogenous microRNAs (miRNAs) that could target the DUX4 transcript, thereby reducing DUX4 expression. So far, he found that H19 and miR-675 reduce DUX4 expression and toxicity. Drs. Harper/Saad seek to develop new therapeutic approaches by targeting or using natural miRNAs such as miR-675. Project aims to create DUX4-targeted miRNA-based gene therapy for FSHD by using miR-675 as a new miRNA-based gene therapy candidate. First by functionally identify every natural human miRNA capable of targeting DUX4 in cell culture. Then by developing a DUX4-targeted miR-675-based gene therapy for FSHD.
There has been tremendous excitement for the therapeutic potential of induced pluripotent stem (iPS) cells in treating genetic diseases. These cells are derived from patients’ skin cells, which are genetically “reprogrammed” to become stem cells, with the ability to develop into muscle.
This project builds on the Perlingeiro lab’s successful studies developing such cell therapies specifically in mouse models of Duchenne and limb-girdle muscular dystrophy (LGMD). The intent of this cell product is to replace diseased muscle with normal functional muscle fibers as well as muscle stem cells, which have the potential to provide long-term therapeutic effect in Duchenne and other devastating types of muscular dystrophies, including FSHD. Because all of the Perlingeiro lab’s work to date has been with Duchenne and LGMD models, it will be essential to understand how effectively cell replacement can address muscle damage due to the distinct mechanism underlying FSHD.
Now that an FSHD mouse model (iDUX4pA) is available that can be induced to produce very low levels of DUX4, resulting in a slow decline in muscle over several months, it will be possible to evaluate the effectiveness of cell therapy in the context of such a relevant muscle damage mechanism. The work proposed in this grant will provide proof of principle for including FSHD in the pipeline for future clinical trials of cell-based regenerative therapies.
Nearly 20 laboratories (including Drs. Mariot’s and Dumonceaux’s) have proposed therapeutic approaches for FSHD, but no one can predict whether any of these approaches will be successful in human patients. It is therefore important to continue to develop new strategies.
This application uses a “decoy” approach, which represents a new conceptual approach in the neuromuscular field. Unlike antisense oligonucleotides (ASO/AO) or siRNAs which target DUX4 messenger (mRNA) prior to the creation of the DUX4 protein, the decoy mechanism of action is to trap the DUX4 protein itself post-RNA translation. The decoy will attach to the DUX4 protein so that it cannot bind to DNA and trigger the downstream toxic effects of DUX4. Notably, this method is independent of the nucleus that produces DUX4 mRNA (which can be one out of 1000 nuclei) allowing the decoy to sequester the DUX4 protein during its cellular journey wherever it occurs.
This decoy strategy may be highly powerful as shown by proof-of-principle studies already performed. The aim of this project is now to validate these results in the FLEx ACTA MCM mice.
FSHD individuals with shorter D4Z4 repeats are reported to be more severely affected, but there is still an unsolved conundrum on different disease manifestation in women and men. Sexual dimorphism in FSHD has been studied among American, Brazilian, Italian, and Dutch FSHD patients. Clinical (e.g., MRI) and neurological data revealed that in these populations, men manifest the disease earlier in their life and are more severely affected than women. The underlying mechanism explaining these noticeable sex differences in disease severity remains yet unsolved and will be the goal of these studies.
Dr. Pakula utilizes fish embryos, which when programmed to synthesize Dux4, develop features that resemble FSHD symptoms. This model is very helpful for studying the mechanism and potential treatments of this disease. By performing analysis of DUX4 binding sites, her team has discovered that, at 12 hours of embryo development, ER-like (estrogen receptor-like) protein interacts with DUX4. The advantage of their model is that DUX4 and estrogen receptor (ER) interaction can be detected at the very early stages of disease development, which is not feasible in humans. The investigators hypothesize that one of the estrogen receptors could help DUX4 reach its binding sites in DNA. Their hypothesis is that in males (having less estrogen than females), DUX4 binds to different DNA regions and regulates different genes, which possibly leads to more severe disease.
Dr. Kunkel and Dr. Pakula, together with Drs. Martha Bulyk and Yuliya Sytnikova from Brigham and Women’s Hospital, who are well established in studying transcription factors and chromatin, will unravel the mechanism of ER-like driven recruitment of the DUX4, which they believe may help to uncover new ways to treat FSHD.
2017 grant awards
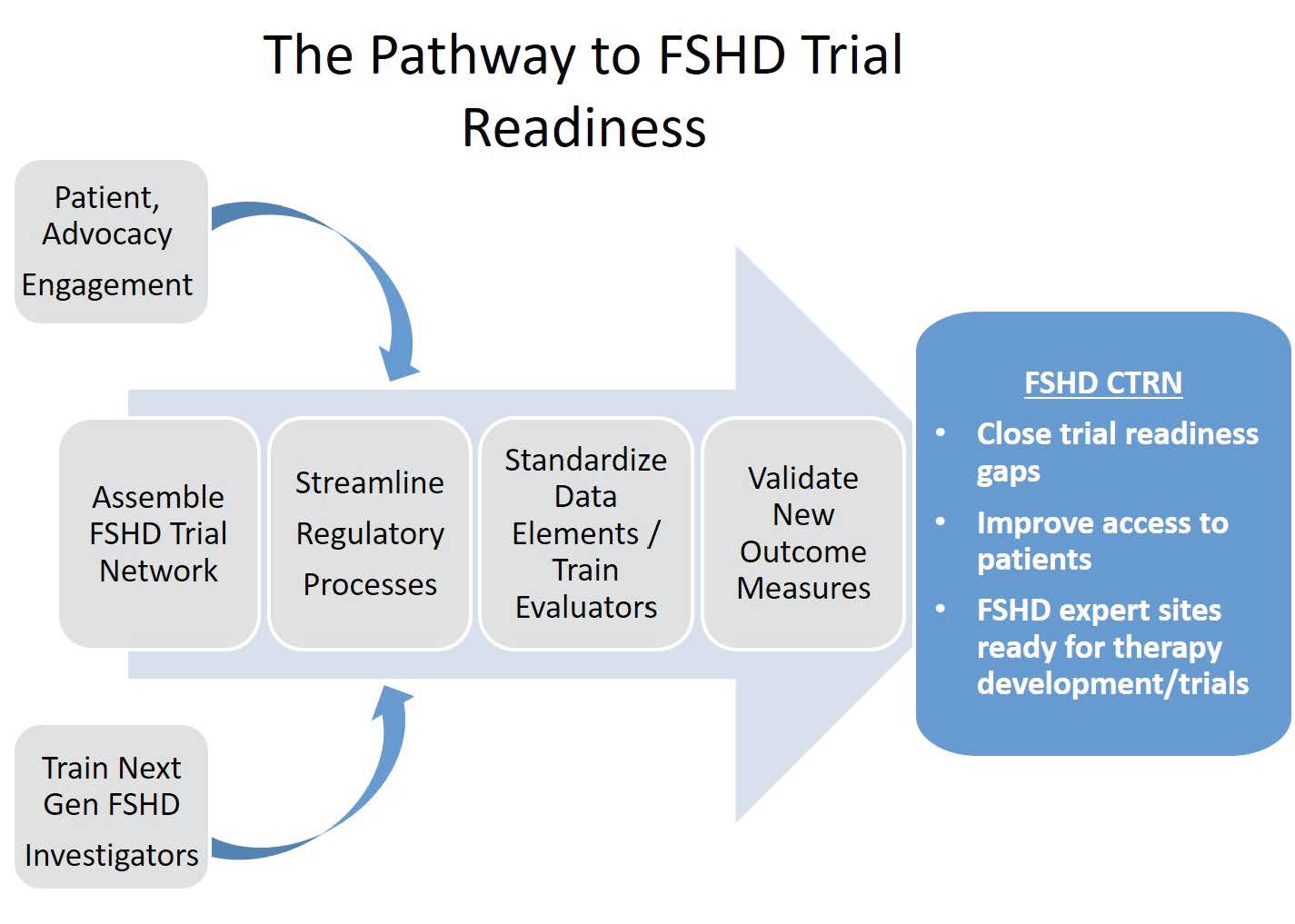
Project Summary. The overall, long term aim of this application is to expedite the development of new therapies for Facioscapulohumeral Muscular Dystrophy (FSHD) by maintaining an FSHD Clinical Trial Research Network (CTRN). Successful clinical trials depend on several factors including: access to and the ability to recruit patients, a precise understanding of the natural history of the disease and the major contributors to disease variability, and reliable outcome measures that are sensitive to change in FSHD. A major hurdle to development of rational trial strategies and validation of outcome measures for FSHD clinical trials is lack of an existing clinical trial network infrastructure with common standard operating procedures (SOPs). We developed the FSHD CTRN to overcome this hurdle by: 1) creating a streamlined system for regulatory /ethical oversight; 2) developing standards for what data is collected and how it is collected; 3) creating a network of well-trained and knowledgeable clinical evaluators – this is absolutely essential for clinical trials; 4) creating a network of trained study coordinators with strong patient engagement, recruitment, and retention skills; 5) ensuring the participation of all major stakeholders; 6) validating new outcome measures for drug registration studies; and 7) training the next generation of FSHD clinical researchers (Figure 1). Not only will the FSHD CTRN help close gaps in trial readiness, but the CTRN also provides a network of sites with a centralized streamlined regulatory process, specific, common expertise in FSHD, and an engaged patient population ready to conduct efficient, high quality clinical trials.
Project Summary. Facioscapulohumeral dystrophy (FSHD) is a complicated disorder. After many decades of study, the FSHD research field now has focused on mis-expression of the DUX4 as a primary insult underlying the disease. DUX4 is toxic to muscle and numerous non-muscle cell types. FSHD symptoms are often variable from person to person, and there may be also variability in severity of symptoms, rate of progression and age at onset, even in families with several affected relatives. Asymmetry is often seen, where a person may have more muscle weakness on one side of the body versus the other. Although DUX4 is toxic, some cells and tissues seem to resist its damaging effects. We hypothesize that FSHD variability and the differential toxicity of DUX4 are linked; it is possible that the toxic effects of DUX4 may be reduced in cells or muscles that are spared in FSHD. However, the mechanisms by which some cells might resist DUX4 damage are unclear. In this proposal, I will investigate my hypothesis that natural microRNAs – which are produced normally in all human cells and help activate natural cellular gene silencing pathways – could reduce DUX4 expression, reduce its toxicity, and potentially slow FSHD progression. In Aim 1, I will continue my investigation of a single miRNA that we identified in a limited candidate screen. This miRNA binds the DUX4 transcript and reduces its translation into DUX4 protein, thereby decreasing its toxicity in cultured cells. This aim represents a proof-of-principle for our second aim, which is focused on identifying the full set of natural miRNAs (humans have 1,881 different ones known to date) that could potentially target DUX4. We would ideally like to tie one or multiple natural miRNAs into FSHD disease progression, but even if we are unable to find evidence for miRNAs acting as natural FSHD modifiers, we propose they could still be used as potential therapeutics if they are capable of binding and reducing DUX4. Specifically, some drugs are known to increase the expression of specific natural microRNAs; thus it may be possible to use drugs to increase the expression of DUX4-targeted miRNAs, thereby reducing the expression of DUX4 so it is no longer toxic.
Project Summary. The D4Z4 repeat array has heterochromatic features in most somatic tissues. As a result, probably due to repeat-mediated epigenetic repression, the transcription factor DUX4 is not, or rarely, expressed in somatic tissues. Individuals with facioscapulohumeral muscular dystrophy (FSHD) present with a partial failure of the epigenetic repression of the D4Z4 repeat array, resulting in DUX4 expression in a subset of muscle nuclei. This failure in epigenetic repression can be caused by contraction of the D4Z4 repeat array to 1-10 units (FSHD1) or by heterozygous mutations in the chromatin modifiers SMCHD1 and DNMT3B (FSHD2). These chromatin modifiers are necessary to establish or maintain the repressed chromatin structure of the D4Z4 repeat array in somatic cells. Our group previously generated a transgenic mouse model carrying a D4Z4 repeat array of 2.5 repeat units. These D4Z4-2.5 mice are a faithful model for some features of FSHD1 since these mice also fail to epigenetically repress DUX4 in somatic cells, leading to the presence of DUX4 protein in sporadic myonuclei.
SMCHD1 encodes a well-conserved protein, but its function is largely unknown. Studies in mice suggest that Smchd1 has roles in the establishment and/or maintenance of DNA methylation, in X chromosome inactivation, and in the regulation of several imprinted and clustered genes. Our group has an ongoing collaboration with Dr. Marnie Blewitt (The Walter and Eliza Hall Institute of Medical Research, Australia), who was involved in an N-ethyl-N-nitrosourea (ENU) mutagenesis screen to identify modifiers of epigenetic reprogramming. Apart from the well-known Smchd1 loss-of-function mutant Smchd1MommeD1, she identified a missense Smchd1 variant, which we now call the Smchd1Fresia variant, which may act as a hypermorphic variant. This is an exciting finding as it suggests that naturally occurring SMCHD1 variants might exist that protect muscle from expressing DUX4.
In this project I will test the hypothesis that specific SMCHD1 variants either increase SMCHD1 activity or lead to increased SMCHD1 expression with consequences for the chromatin structure of the D4Z4 repeat array and for DUX4 expression. In Specific Aim 1, I will determine the functional consequences of the Smchd1Fresia variant at the chromatin and expression level of the D4Z4 repeat array in vivo using our transgenic D4Z4- 2.5 mice. In Specific Aim 2, I will determine the effect of the Smchd1Fresia variant and five SMCHD1 variants that may act as hypermorphic alleles in muscle cell cultures. In Specific Aim 3, I will search for novel potential hypermorphic SMCHD1 variants in our extensive and well characterized biorepository.
Project Summary. Facioscapulohumeral Dystrophy (FSHD) is a human specific dominant genetic disease caused by the contraction of a D4Z4 repeat array at chromosome 4q35 and a permissive epigenetic environment. These genetic and epigenetic circumstances lead to muscular dystrophy in patients with highly variable rates of muscle group penetration and progression. Recent advances in the FSHD field have shown that each D4Z4 repeat contains a gene called DUX4, and that only the most distal repeat in the contracted array is capable of producing translatable pathogenic transcripts, called DUX4-fl. Recently, Dr. Peter Jones was able to generate the first viable and fertile line of inducible DUX4-fl transgenic mice, referred to as FLExDux4+/-; ACTA-MCM+/-. This mouse model is now being leveraged by us and other labs to examine novel drugs that may have efficacy for FSHD patients. In Dr. Dean Burkin’s lab we have previously performed a large-scale drug screen to identify small molecule enhancers of ITGA7, the gene encoding alpha7 lntegrin. In studying these “hit” compounds in other muscular dystrophy mouse models, we observed that a select agent gave us a large improvement in muscle regeneration along with the expected increase in alpha7 lntegrin. This regeneration occurred in the absence of telomere length shortening. We have gone on to perform preliminary treatments in the FLExDux4+/-; ACTA-MCM+/- mouse model, and while we see no decrease in DUX4-fl target gene expression or activity, we do find a large increase in ex vivo muscle force production. We hypothesize that Stryka-001 treatment of the tamoxifen treated FLExDux4+/-:ACTA-MCM+/- FSHD-like mouse model will improve muscle regeneration and recovery after DUX4-fl induced muscle insult. If successful, this technology will have immediate treatment implications for FSHD patients and will be extremely useful in combination with other upcoming therapeutic interventions targeting DUX4-fl.
Project Summary. Facioscapulohumeral muscular dystrophy (FSHD) is one of the most prevalent neuromuscular disorders. Due to incomplete understanding of its molecular pathogenesis, no treatment is currently available. The disease is caused by aberrant expression of the double homeobox 4 (DUX4) gene encoding for a transcription activator normally silent in skeletal muscle. In FSHD, ectopic DUX4 expression activates a pro-apoptotic transcriptional program leading to muscle cell loss and degeneration. While blocking DUX4-induced toxicity would be a plausible therapeutic option, the mechanism through which DUX4 triggers cell death is poorly understood and no regulator of DUX4 activity is currently known. Hence, the identification of factors able to block DUX4-activated toxicity is crucial when considering future drug design.
We identified a novel molecule able to block DUX4 activity.
We plan to address the following questions: 1. Which are the molecular determinants of DUX4-inhibitor interaction? 2. Can the inhibitor be used for therapeutic purposes?
Our project will provide a better understanding of DUX4 mechanism of action and how its toxic activity could be blocked for the treatment of FSHD.
Project Summary. Facioscapulohumeral muscular dystrophy (FSHD) is a common muscle wasting diseases, caused by a combination of genetic and epigenetic abnormalities in the D4Z4 marcosatellite repeat array in the subtelomere of chromosome 4 at 4q35. The most common form, FSHD1, is linked to contraction of D4Z4 array from the 11-100 repeats in unaffected individuals, to less than 10. In the other 5% of cases (FSHD2), the D4Z4 region is un-contracted. Both forms are associated with epigenetic changes to the region such as DNA hypomethylation and loss of heterochromatic histone marks, which renders the region permissive to transcription. If such a hypomethylated D4Z4 array is present on a permissive 4qA allele supplying a polyA signal, a stabilised transcript from the terminal D4Z4 repeat for a transcription factor called DUX4 is made. When ectopically expressed in skeletal muscle, DUX4 disrupts the transcriptional networks of muscle cells and has a cytotoxic effect. However, molecular drivers of FSHD pathology remain poorly understood. Upon injury, healthy muscle, in cooperation with the immune system, activates a complex repair program that involves activation of muscle-progenitor (satellite) cells that proliferate and differentiate to repair damage. These processes in FSHD are mis-regulated, which leads to an abnormal inflammatory response, ineffective repair and myofibre atrophy. Understanding the failure of FSHD muscle to activate effectively the muscle repair program could be important in developing novel therapeutic strategies.
In work partially funded by the FSH Society Shack Family and Friends research grant FSHS-82013-06), we have recently completed an extensive RNA-seq transcriptomics analysis of myogenic differentiation of immortalised and primary myoblasts isolated from FSHD patients alongside matched controls (Banerji C.R.S, Panamarova M., Hebaishi H., White R.B., Relaix F., Severini S. and Zammit P.S. (2017). PAX7 target genes are globally repressed in FSHD skeletal muscle. Nature Communications 8: 2152 (10.1038/s41467-017-01200-4). Multivariate regression analysis revealed 180 genes strongly associated with FSHD in every dataset analysed. Gene Set Enrichment analysis of these 180 genes revealed that the target genes of a transcription factor central to macrophage-coordinated muscle repair were significantly repressed in all FSHD cell lines. However, the effects of suppression of this transcription factor on muscle repair in FSHD, is currently unknown.
This research aims to determine the role of this transcription factor in FSHD, which could help augment muscle repair in FSHD to ameliorate muscle wasting. An overarching aim of this project is to better understand the interplay between muscle repair and the immune system in FSHD.
Project Summary. To move forward in clinical trial readiness in FSHD, the identification of biomarkers of activity and progression is required to help assessing the efficacy of a treatment in a slowly progressing disease. Selective and targeted approaches are advisable in this disorder, and a correlation of molecular findings with other measures of disease activity and progression is needed to reduce the variability of the results. We developed an original approach that combines muscle imaging, microdialysis and proteomic analysis to identify and track the pathological processes taking place in single muscles. This approach, which consists in the proteomic analysis of interstitial fluid obtained from muscles with different MRI features (i.e., normal muscles vs. muscles showing signs of early involvement) in the same FSHD patients and controls, allows the contextualization of the molecular results in the frame of the comprehensive and sensitive assessment provided by MRI. Preliminary evidences on already collected samples support the feasibility of the analysis. After the discovery phase, we also plan to develop a sensitive and robust proteomic workflow to verify and enhance sensitivity of detection and quantification of proteins/peptides. Accurate masses of targeted peptides from proteins that were differentially expressed in the microdialysates will be analyzed in high-resolution LC-PRM (Liquid Chromatography-Parallel Reaction Monitoring)-mass spectrometry (MS) analysis mode, which is considered the method of choice for the verification step in biomarker discovery using MS. Proteomic protocols will be developed and tested for biomarker discovery also in serum of the same FSHD patients that underwent microdialysis.
The results of our study could provide information valuable for the discovery and characterization of novel tissue and circulating biomarkers with a comprehensive approach, as well as preliminary evidence for the application of an innovative technique in FSHD and potentially other neuromuscular disorders. Getting further insights into disease pathophysiology through the identification of biochemical pathways dysregulated in FSHD muscles could help in the development of new targeted therapies.
Project Summary: Facioscapulohumeral dystrophy (FSHD) is a common but unique form of muscular dystrophy requiring multiple factors to create a ‘permissive’ state for disease manifestation. Over recent years, several genetic (DUX4) and epigenetic (hypo-methylation) factors have been linked to FSHD pathogenesis; however, it has become clear that the field has not elucidated all factors required for disease manifestation. Mounting clinical evidence suggests the existence of modifier genes with the capacity to regulate DUX4 transcript and/or protein function. Recent advances in genome-editing technologies proposed for use in this project now should enable us to uncover these remaining missing links. Through the systematic introduction of loss-of-function mutations into genomic DNA, we can interrogate the genome for answers that may explain the phenotypic variability between patients, as well as the non-penetrant effects of DUX4 in some individuals. In this project, we propose a targeted genome-scale knock-out screen to identify genes that can reduce the phenotypic impact of DUX4 expression when inactivated. We hypothesize that there exists gene targets of DUX4 whose loss will render DUX4 unable to trigger a dysregulated cascade of gene expression, thus abrogating its toxicity. These candidates likely serve as genetic modifiers of FSHD, and will be readily identified by downstream sequencing and computational analysis for detection of CRISPR target genes enriched within these DUX4 ‘resistant’ cell populations. This will allow the generation of a complete list of gene candidates with the potential to influence the pathogenic outcomes associated with DUX4 misexpression. Identified gene hits will be cross-referenced to our whole-genome sequencing data of nonmanifesting carriers to search for sequence variants that may enable us to narrow down promising candidates for functional follow up studies. Validation of candidate modifier genes will be performed in our established zebrafish model of FSHD for rescue of phenotype to confirm functional significance. Additionally, we will revert to our repository of FSHD patient cells to genome edit our candidate genes under these permissive allelic conditions, and subsequently measure changes in known FSHD biomarker expression. FSHD is a challenging disease whose remaining unanswered questions cannot be accomplished alone. Hence, our proposal involves a multi-institute collaboration, bringing together a wealth of patient resources (Wellstone Center), the latest in genomic technology (Broad Institute), and a well-established animal model of FSHD (Boston Children’s Hospital). Not only will the identification of these modifier genes for DUX4 resistance provide valuable insights into FSHD disease pathogenesis, but they will also present as solid leads that can be directly targeted for therapeutic intervention in humans with FSHD.
Project Summary: Facioscapulohumeral muscular dystrophy (FSHD) has two types, FSHD1 and FSHD2. The causative gene of FSHD1 and FSHD2 is DUX4. The reasons that DUX4 is expressed in FSHD1 and FSHD2 are contraction of the D4Z4 macrosatellite repeat unit and mutations in SMCHD1, respectively, combined with 4qA allele carrying the DUX4 polyadenylation site. In addition, SMCHD1 modifies disease severity in families affected by FSHD1. Here, to understand the molecular mechanisms to express DUX4, I seek to identify how SMCHD1 is involved in DUX4 regulation (Aim1 and Aim2). Moreover, I seek to elucidate the role of Smchd1 in FSHD1 model mice (Aim3). Aim1. Identification of the molecular mechanism used by the mini-SMCHD1 to derepress DUX4 expression. Mini-SMCHD1 (Exon1-9.41-48) de-represses DUX4 expression. To clarify the mechanisms of the increased DUX4 expression by mini-SMCHD1 in FSHD1 myoblasts, I test whether the mini SMCHD1 decreases the amount of endogenous SMCHD1 protein and/or whether the mini SMCHD1 binds to the D4Z4 region and displace the endogenous SMCHD1. Aim2. Identification of the molecular mechanism of SMCHD1 cleavage. There could be full-length and cleavage fragment bands on endogenous SMCHD1 in control and FSHD1 muscle cells. My hypothesis is that SMCHD1 that lacks putative cleavage sites could be more stable than endogenous SMCHD1 and it has better ability to repress DUX4 expression. To test this, I will first identify which sequence is recognized by which protease with bioinformatics and molecular biology tools. Next, I will investigate whether the protease and its recognition sites involved in the cleavage of SMCHD1 could be a novel target for therapeutic intervention. Aim3. Identification of the role of Smchd1 in FSHD1 model mice. Decreased SMCHD1 level de-represses DUX4 expression in FSHD1 myoblasts. To investigate whether Smchd1 could affect DUX4 expression in FSHD1 model mice, I will compare DUX4 expression in Smchd1 conditional knockout mice (D4Z4-2.5; Myf5Cre/+; Smchd1flox/flox) with that in control mice (D4Z4-2.5; Myf5+/+; Smchd1flox/flox) both under injury condition as well as normal mature muscle.
Project Summary: For FSHD the current model suggests that a shortened D4Z4, subsequent DNA hypomethylation at a permissive 4qA allele induce expression of the DUX4 transcript that in turn activate other genes leading to the muscle-specific phenotype. For a small subset of patients (approximately 5%, FSHD2), the clinical phenotype is identical but appears without D4Z4 array contraction. However, most of these patients display a profound D4Z4 hypomethylation linked to some of them to mutations in the SMCHD1 (Structural Maintenance of Chromosomal Hinge Domain Containing) gene. Epigenetic alterations are thus closely associated to FSHD but the underlying mechanisms remain unclear and the functions of the SMCHD1 protein in the regulation of D4Z4 remains partly understood. SMCHD1 is a large 230 kDa non-canonical member of the SMC family of chromosomal proteins. The main conserved domains are the carboxy-terminal SMC hinge domain flanked by short coil-coiled regions, the amino-terminal GHKL ATPase domain and a region with weak homology to the Bromo-adjacent homology (BAH) domain near the ATPase domain. SMCHD1 is able to homodimerize through the SMC hinge domain and is preferentially loaded on H3K9m3- enriched chromatin. In the mouse, Smchd1 is mainly characterized for its implication in X chromosome inactivation. Smchd1 is also involved in silencing of repetitive DNA sequences, regulation of clustered imprinted genes and of the monoallelically expressed Protocadherin genes cluster. SMCHD1 has also been found at telomeres with a direct correlation between telomere length and SMCHD1 enrichment. However, its precise role at telomeres is unknown. The aim of this project is to understand the role of SMCHD1 in chromatin regulation and DNA methylation during muscular differentiation to uncover how this protein contributes to the physiopathomechanisms underlying the FSHD disease. To this aim, we will use induced pluripotent stem cells from patients affected with FSHD1 and FSHD2 carrying different SMCHD1 mutation. We have developed in the team a strategy for the production of skeletal muscle cells from hiPSCs (myoblastes and myotubes) which will be use to monitor the expression of genes dysregulated during differentiation and to determine the profile of SMCHD1 binding to chromatin. The goal of this project is to identify pathways dysregulated in the disease in order to get further insights into the disease pathomechanism.
Project Summary: Facioscapulohumeral muscular dystrophy (FSHD) a genetically dominant progressive muscular dystrophy associated with derepression of the DUX4 gene. One of the major current roadblocks to FSHD basic research and therapeutic testing is the lack of a suitable animal model, with existing attempts either being too severe or lacking a muscle disease entirely. We have developed a new transgenic mouse with tissue-specific and titratable DUX4 expression that shows skeletal muscle disease and this application proposes to develop this into an animal model suitable for studying the role of the DUX4 protein in both skeletal muscle fibers and in the stem cells for skeletal muscle. This work enables studying skeletal muscle pathology due to the DUX4 gene in vivo, and has the potential to enable testing of therapies for FSHD based in inhibiting the DUX4 protein or RNA.
Project Summary: Facioscapulohumeral muscular dystrophy (FSHD) is a developmental disorder in which DUX4 expression is not silenced in early myogenic events. A proper model of early human myogenesis could elucidate a new pathogenic mechanism for FSHD. During the last two generous funding supports from the FSH Society (2012 and 2014), we have established multiple FSHD and healthy control human induced pluripotent stem cell lines (hiPSCs). In addition, the Lee lab has developed a novel ‘chemical compound-based’ skeletal muscle differentiation methodology without using overexpression of myogenic transcription factors, animal products, or even recombinant proteins (published in 2016, Choi et al., Cell Reports). This new protocol is relatively fast (~ 30 days) and faithfully follows in vivo myogenesis. For example, our genetic reporter system (MESOGENIN1::eGFP, as a marker for pre-somite stage) shows that over 80% of the differentiating cells are undergoing the somite stage, suggesting that our protocol is indeed mimicking developmental myogenesis. The results from our previous funding support (2014 funding from the FSH Society) indicate that the SSEA3+ undifferentiated iPSCs, MESOGENIN1::GFP+ somite cells, and even NCAM+/HNK1- myoblasts cells may not be the best cell type to discern the effects of DUX4 and/or FSHD-related transcriptional discrepancies (please refer to our research progress report in ATTACHMENT IV). In FSHD patients, it is unclear which cell types express DUX4, and it is extremely difficult to find any DUX4 immunoreactive cells in human primary myoblast cultures. A very recent study from the Zammit group shows that Dux4 is transiently expressed during skeletal muscle regeneration, Dux4 maintains Pax7 expression through transcriptional activation of target genes, and Dux4 induces signatures of a stem-cell-like and less-differentiated state. These data lead us to hypothesize that DUX4 can be expressed in PAX7 expressing cells of FSHD iPSCs, or at least PAX7 expressing cells should be the correct cell type to study FSHD molecular pathogenesis. My group has developed a strategy to generate ‘knock-in’ PAX7::GFP reporter lines, and we have established multiple PAX7::GFP reporter human iPSCs. We will continue our efforts to generate PAX7::GFP FSHD-hiPSC and control-hiPSC lines (three genotypes for each) to isolate putative skeletal muscle stem/progenitor cells, followed by detailed cellular and molecular analysis. These approaches can lead to new insights on FSHD disease mechanisms, and the newly developed cell lines can be shared with other research groups for future in vitro and in vivo studies.
Project Summary: FSHD is one of the most common muscular dystrophies and so far there is no curative or preventive treatment. It is characterized by a loss of repressive epigenetic marks within the D4Z4 array, leading to chromatin relaxation and, when associated with a permissive chromosome 4, to the expression of the normally silenced DUX4 protein whose ORF is present in each D4Z4 repeat. DUX4 is a transcription factor resulting in a poison protein through induction of downstream genes, which might play a major role in FSHD onset/progression.
DUX4 is often described as toxic for muscle cells in FSHD but cell loss mechanism driven by DUX4 expression remains largely unknown. In the literature, several articles have already investigated DUX4-dependent cell death mechanisms in vitro and in vivo, but focusing on apoptosis pathways. However, whereas FSHD mainly involves cells dying with necrotic morphology in patients biopsies, the necrotic death pathway has never been investigated. Our goal is to investigate necrotic mechanisms in this pathology.
This work will allow a better understanding of FSHD pathophysiology, may explain the link between DUX4 expression and FSHD pathophysiology and may help to define new therapeutic targets.
Project Summary: Facioscapulohumeral muscular dystrophy (FSHD) is a currently untreatable genetic disease whereby patients suffer from progressive muscle weakness. The genetic alteration causing FSHD has been mapped to the D4Z4 macrosatellite repeats at chromosome 4q35.2. In healthy individuals, this region has 11-100 copies of D4Z4 and remains intrinsically silenced. However, in FSHD patients, D4Z4 repeats were contracted to be less than 11 copies, leading to epigenetic de-repression of D4Z4 and transcriptional activation of double homeobox 4 (DUX4), a transcription factor residing within D4Z4. Although these phenomena have been well-documented, the factors and mechanisms contributing to DUX4/D4Z4 misregulation still remain largely unknown. Recently, Dux4 was shown to possess transcriptional transactivities and positively regulate germline development and immune response genes that potentially contribute to FSHD pathogenesis. However, upon Dux4 over-expression in myoblasts, a subset of Dux4 direct target genes were found downregulated, underscoring the possibility that Dux4 may participate in functionally distinct complexes to regulate gene expression. To probe the factors regulating DUX4/D4Z4 and dissect the functions of Dux4, we propose to reconstruct the epigenetic landscape of the D4Z4 locus, screen for transcription factors that activate DUX4 expression, and map the interactome of Dux4 in order to uncover the potential regulators of D4Z4 repression, DUX4 expression, and Dux4 function on chromatin, respectively. Specifically, proteins bound to the D4Z4 locus will be purified and identified using proteomics of isolated segmented chromatin (PICh). Candidate transcription factors that potentially regulate DUX4 expression will be screened using a focused shRNA library. Dux4-interactors will be identified by chromatin immunoprecipitation or chromatography fractionation of DUX4 protein followed by mass spectrometry analyses. Functional validation following these target discovery approaches will be performed in normal and FSHD skeletal myotubes as well as patient biopsies. Collectively, these attempts will elucidate the molecular mechanisms underlying FSHD pathogenesis and reveal potential targets for therapeutic interventions to treat FSHD.
Project Summary: Muscle wasting is one of the biggest challenges in neuromuscular disorders. Myostatin being a negative regulator of muscle mass, its down-regulation has been seen as a promising tool to counterbalance this muscle wasting and at least 6 anti-myostatin molecules have been developed by pharmaceutical companies. However, so far, the clinical trials have been very disappointing and clinical endpoints have been barely reached. These results are surprising since during the phase 1 trials on healthy volunteers, an improvement of muscle mass was observed.
Several hypotheses have been proposed among them the poor efficacy of the anti-myostatin molecules or the specificity of the drugs themselves. In our study we are investigating another possibility based on the expression levels of several effectors of the myostatin pathway. Our experiments indicate that patient’ stratification (based on the expression of these effectors) might be useful to determine patient eligibility.
The funding provided by the FSH society will help us to finish performing the experiments which may be of importance for neuromuscular patients, and FSHD patients in particular, and may deeply impact future and current clinical trials using myostatin inhibitors.
2016 grant awards
In response to a request from the FSH Society, NDRI proposes to develop and implement a resource to recover surgical and post mortem human bio specimens and distribute them to approved investigators. This resource will utilize NDRI’s experience, expertise and established systems to expand and enhance the type, number and quality of human tissues available to the FSH research community. lt is proposed that NDRI’s Private Donor Program will collaborate with FSH to recover and distribute tissues from patients who participate in the FSH Registry and who have provided consent for the recovery of tissues and organs for research. In addition to providing all resources required to recover tissues post mortem and from surgical procedures, NDRI will provide informational materials to the FSH Society for distribution to potential registry participants, as well as IRS-approved templates for obtaining informed consent from patients and authorization to donate from family decision makers.
Pathogenesis in Facioscapulohumeral muscular dystrophy (FSHD) appears to be due to aberrant expression, particularly in skeletal muscle nuclei, of the full-length isoform of DUX4 (DUX4-FL). DUX4-FL had been shown to induce toxicity by ectopic expression and can lead to aberrant expression of DUX4-FL target genes including ubiquitin ligases, ubiquitin binding proteins, and RNA processing genes as well as germline and stem cell genes (1-6). We and another group identified disturbed proteostasis as a possible mechanism for DUX4-mediated pathology (7, 8). Dysregulation of proteostasis can interfere with normal cellular functions, cause stress or immune responses, and lead to disease. Abnormal RNA or protein accumulation has been implicated in a number of diseases including amyotrophic lateral sclerosis (ALS), inclusion body myositis (IBM), and other myopathies (9-14). DUX4 expression also inhibits nonsense-mediated decay (NMD) (8), which can lead to abnormal expression, processing, or accumulation of RNAs and protein, including DUX4 itself. We discovered that DUX4-FL, but not DUX4-S, inhibits protein turnover and leads to abnormal ubiquitin expression and nuclear aggregation of TDP-43 (TAR DNA-binding protein 43), one of the aggregation-prone and RNA/DNA binding proteins previously associated with ALS and IBM (7). Importantly, the abnormal deposition of ubiquitinated protein and nuclear aggregation of TDP-43 were observed when DUX4-FL was expressed from its endogenous promoter, as well as when it was exogenously expressed. For this project, we hypothesized that DUX4-FL expression would induce progressive impairment of the ubiquitin-proteasome system (UPS). We proposed, therefore, to identify mechanisms that underlie the DUX4-FL-induced dysregulation of proteostasis and protein Page 3 of 5 aggregation as a step to understanding pathogenesis and developing therapeutic strategies for FSHD. The two specific aims are to 1: Identify the mechanisms by which DUX4-FL inhibits protein turnover and 2: Determine if FSHD muscle tissues show signs of disturbed proteostasis. As in one-year progress report and request for funding extension, we have made significant progress towards accomplishing both aims.
Facioscapulohumeral muscular dystrophy (FSHD) is an adult-onset, autosomal dominant disorder initially characterised by wasting of facial muscles and upper body musculature. Disease can progress to affect muscles of the lower extremities and severely impair quality of life. Over 95% of FSHD cases are classed as FSHD1, caused by contraction to less than 11 units of the D4Z4 microsatellite repeat, on the subtelomeric region of chromosome 4. At least one D4Z4 unit is required to cause FSHD however, and only when inherited with a specific polymorphism on the distal end of chromosome 4 (e.g. 4qA161). Each D4Z4 unit contains an open reading frame for the double homeobox 4 (DUX4) retrogene, with specific 4qA haplotypes providing a polyadenylation signal for DUX4 transcripts generated by the last D4Z4 unit. This permissive chromosomal configuration generates stable DUX4 transcripts and FSHD is caused by a toxic gain-of-function of DUX4. FSHD2 is caused by mutation in genes responsible for methylation at D4Z4, with the resulting hypomethylation again causing DUX4 expression, but without contraction at D4Z4. FSHD myoblasts are particularly sensitive to oxidative stress. Thus treatment by anti-oxidants has been explored as a therapy. A recent clinical trial (clinicaltrials.gov number: NCT01596803) administered vitamin E, vitamin C, zinc, and selenomethionine to FSHD patients for 17 weeks to enhance anti-oxidant defense and reduce oxidative stress. They reported improved maximal voluntary contraction and endurance limit time in quadriceps muscle of the treated patients, but no effects on the two-minute walking test (Passerieux et al. 2014 – doi:10.1016/j.freeradbiomed.2014.09.014). To identify pathways that lead to compromised muscle function, we have performed RNA-Seq on cell lines derived from FSHD patients in a high-frequency time course of genome wide gene expression during myogenic differentiation (funded by the FSHSociety). Using mathematical methodologies with optimised network theoretic tools on this gene expression dataset, will have revealed molecular mechanisms of myogenesis in FSHD. This analysis of our RNA-Seq time course data has led to a number of novel insights into FSHD molecular mechanisms, particularly implicating critical mediators of oxidative stress, mitochondrial biogenesis, the TCA cycle and myogenic progression, as perturbed in FSHD. Our RNA-Seq data indicated suppression of mitochondrial biogenesis during FSHD myogenesis, and mitochondrial dysfunction has been reported in FSHD, indicating that activation of this pathway could provide a therapeutic strategy. For rapid translation to the patient/clinic, we have investigated nutritional supplements that target this pathway and found that several improve myogenesis of FSHD patient derived cells. In this project, we will screen several more nutritional supplements and select the most promising for testing in a wide range of different FSHD patient derived myoblasts. We will also test selected nutritional supplements on myoblasts expressing DUX4, to determine their effectiveness at ameliorating the drastic phenotype elicited by DUX4. Effects of nutritional supplements on signaling pathways that are perturbed in FSHD will also be examined to better understand their mechanism of action. By analyzing modifiers of these pathways we hope to improve our understanding of the molecular defects in FSHD and how best to modify them to maximize patient benefit. Ultimately the aim is translation of such an approach to a clinical trial setting, and as we focus on nutritional supplements, it is likely that such translation could be rapid.
Facioscapulohumeral muscular dystrophy (FSHD) is believed to be caused by the aberrant expression of double homeobox protein 4 (DUX4) due to epigenetic changes at chromosome 4q35 region. Antisense oligonucleotide (AON) therapy is a promising strategy to eliminate pathogenic gene product, such as DUX4 mRNA, in cells. In this study, we will investigate one of promising AON compounds called LNA gapmer for its efficacy in reducing DUX4 in cell culture and in a mouse model of FSHD. The findings will allow us to evaluate this compound as a potential treatment for FSHD. Antisense therapy shows promise for treating an array of disorders, however, several problems associated with AONs yet to be improved, including 1) difficult in systemic drug delivery because these AONs could not easily cross the lipid bilayer of cells; 2) harmful off-target effects and immune responses via toll-like receptors; 3) low stability due to degradation by intracellular and extracellular nucleases. Considering these challenges, locked nucleic acids (LNAs) show exceptional thermal stability, impose significant protection against nucleolytic degradation and have a high binding affinity. Importantly, LNA can be systemically delivered in vivo. Modifications to the LNA gapmer chemistry have also shown great success and allow RNase H-mediated cleavage to degrade target RNAs. In our preliminary study, we designed LNA gapmers targeting DUX4 and successfully knocked down DUX4 mRNA in immortalized FSHD myoblasts. The goal of this study is to further characterize LNA gapmers for its efficacy and safety in vitro and in vivo. The studies will be conducted by two highly experienced investigators with complementary expertise in the field. Dr. Toshifumi Yokota who is an expert in AON therapy and has designed and generated the in vitro preliminary data in collaboration with Dr. Chen. Dr. Yi-Wen Chen has extensive experience in FSHD and FSHD mouse models. Dr. Yokota will be in charge of designing the LNA gapmers and performing in vitro studies using immortalized FSHD myoblasts as proposed in Aim 1. Dr. Chen will be in charge of performing in vivo studies using a new mouse model of FSHD to determine the efficacy of the LNA gapmers in vivo as proposed in Aim 2. The two investigators have been closely working together to develop this study and will keep the collaborative nature of work during the funding period. The goal is to carefully characterize the LNA gapmers that target DUX4 and identify those with the highes efficacy and specificity for treatment development. There is no effective treatment for FSHD, however, aberrant expression of DUX4 is known to cause this disorder. The proposed studies will study an effective antisense oligonucleotide strategy to target DUX4 and reduce its expression. LNA has been studied in vitro and in vivo for efficacy and safety extensively. We have generated preliminary data to show effective DUX4 knockdown in FSHD myoblasts. This collaborative study will characterize the LNA Gapmers against DUX4 further as potential therapeutics for FSHD.
Facioscapulohumeral muscular dystrophy (FSHD) is characterized by extreme variability in symptoms with females being less severely affected than males and presenting a higher proportion of asymptomatic carriers. Thus far, gender factors involved in the disease have not been identified. Recent data from our group demonstrate that estrogens improve in vitro the differentiation ability of myoblasts from FSHD patients without affecting cell proliferation or survival. Specifically, estrogens counteract the muscle differentiation impairment caused by the homeobox protein DUX4, the best FSHD candidate gene. We further demonstrated that estrogen receptor beta (ERβ), present in female and male individuals, is involved in this activity by displacing DUX4 from the nucleus and impairing its transcriptional function. Importantly, both 17β-estradiol, the predominant female hormone regarding estrogenic activity, as well as 5α-Androstane-3β,17β-diol (3β-diol), a natural endogenous ligand of ERβ present in males, can promote this ERβ-mediated activity. The present project aims to confirm these data in vivo by analyzing the effect of estrogen on the ability of transplanted human muscle-derived cells to participate in the regeneration of injured muscle in immune-deficient mice. The choice of this model is based on the following reasons: 1.) The role of muscle differentiation defects in the pathophysiology of FSHD is still controversial. Conversely, muscle degeneration with fatty replacement in humans has been shown as well as impaired regeneration in FSHD mouse models; 2.) the ability of FHSD muscle-precursor cells as mesoangioblasts, or of FSHD myoblasts to differentiate into skeletal muscle in immunodeficient mice has been previously reported; 3.) it has been recently reported an innovative approach for the generation of a mature skeletal muscle based on a hydrogel/growth factor scaffold. Based on these data, we reasoned that the model of muscle regeneration is a suitable model to test the beneficial effect of estrogen in vivo. Given the low proliferative potential of myoblasts, we will use muscle-precursor cells (perivascular cells). Specifically, we propose to transplant human perivascular cells expressing exogenous or endogenous DUX4 in muscle-ablated mice and analyze their ability to form myotubes depending on the levels and/or activity of estrogens. The project includes three main tasks: 1.) analyze the effect of estrogen on transplanted human muscle-precursor cells. This task will establish the experimental conditions for further analyses: – test the growth of PVCs dependent on the gender and the levels of different estrogenic compounds; – test the best conditions for growth of exogenous DUX4-expressing PVCs. 2.) analyze the regeneration ability of DUX4Cherry-PVCs depending on the levels/activity of estrogens. This task is the core of the project, aiming to analyze the regeneration ability of DUX4-expressing cells depending on the levels/activity of estrogen. Different groups of animals will be used based on gender difference and/or estrogen levels or activity (using specific estrogen antagonist able to bind ERβ and to compete with natural endogenous ligands). 3.) analyze the regeneration ability of FSHD-derived PVCs. PVCs derived from FSHD patients will be subjected to the same protocol of transplantation and their myofiberformation ability tested. Given the preciousness of FSHD muscle biopsy, based on the results of the previous task, FSHD-derived PVCs will be challenged in the most efficient conditions. This project will ascertain the in vivo role of estrogens towards FSHD, particularly on the regeneration ability of DUX4-expressing PVCs. The success of this project will support the estrogen as one of the factors underlying FSHD gender differences and will establish their protective function against this disease. Most importantly, these data might open the venue to therapeutic intervention in FSHD patients.
Specific Aims: Facioscapulohumeral muscular dystrophy (FSHD) is caused by the misexpression of the germline transcription factor DUX4 in muscle cells. Several mechanisms have been proposed to explain DUX4-induced myotoxicity, including activation of apoptotic pathways, perturbed proteostasis and protein aggregation, among others (Wallace et al., 2011; Wallace et al., 2012; Tassin et al., 2013; Homma et al., 2015). We recently observed profound inhibition of an essential RNA quality control mechanism – nonsense-mediated RNA decay (NMD) – following DUX4 expression that could potentially account for several aspects of FSHD biology (Feng et al., 2015). Temporal analysis of DUX4 expression in human myoblasts revealed proteolytic degradation of core NMD factors upon DUX4 expression and concomitant inhibition of NMD. Following NMD inhibition, the DUX4-expressing cells upregulated various protein folding stress response pathways, leading us to hypothesize that DUX4-induced NMD inhibition could allow synthesis of aberrant protein products and cause proteotoxicity and cell death. In support of this hypothesis, pilot quantitative mass spectrometry studies detected truncated proteins as well as a small number of novel peptides derived from NMD targets in the DUX4- expressing cells. To rigorously test whether NMD inhibition drives proteotoxicity and contributes to DUX4-induced cell death, we sought to identify the mechanism of NMD inhibition by DUX4 (Specific Aim 1) and to determine the contribution of inefficient NMD to DUX4 toxicity (Specific Aim 2).
Specific Aims: We recently published the first, and still only, report utilizing the CRISPR/Cas system for reducing or eliminating DUX4-fl expression as an avenue to an FSHD treatment [Himeda et al. “CRISPR/dCas9-mediated transcriptional inhibition ameliorates the epigenetic dysregulation at D4Z4 and represses DUX4-fl in FSH muscular dystrophy. 2015. Molecular Therapy, (In Press)]. Importantly, in this work we showed that it is in fact feasible to design sgRNAs that target Cas9 to the 4q35 D4Z4 in primary human myogenic cells. The next phase is to move to an in vivo system and ask if we can target the D4Z4/DUX4 in an animal model; however, due to off target concerns, it is also vital to be targeting mature human muscle fibers. Fortunately, we have been collaborating with Dr. Bob Bloch and his colleagues at the University of Maryland School of Medicine on their humanized mouse models for FSHD. Recently, they published the first study on these mice and show that their technique produces spectacular engraftment and development of mature human muscle fibers, with minimal contamination by mouse myonuclei [Sakellariou et al. “Neuromuscular electrical stimulation promotes development in mice of mature human muscle from immortalized human myoblasts” 2016. Skeletal Muscle (In Press)]. Thus, the timing works out perfectly for us to continue our collaboration and merge our CRISPR/Cas studies with their mouse model. In this collaborative grant the Jones lab at UMMS will develop new sgRNAs targeting the D4Z4/DUX4 region that are compatible with the smaller Cas9s which fit into AAV. The Bloch lab at UMSOM will generate humanized FSHD mice, inject these CRISPR AAVs, and assess their effects on muscle physiology and morphology. The Jones lab will then analyze the expression of DUX4, downstream targets, and off targets. This project will test if AAV-mediated CRISPR technology is indeed a viable approach to treating FSHD.
Specific Aims: The DUX proteins are known as transcription factors and until now functional studies from several laboratories only focused on that perspective. Intriguingly, the transcriptional target genes of DUX4 identified to this date cannot explain the cytoplasmic alterations observed in FSHD muscle sections. In a search for DUX4/4c protein partners we have unexpectedly identified several classes of cytoplasmic or nucleo-cytoplasmic proteins. The functionality of these interactions was underscored by the observation of DUX4/4c cytoplasmic localization upon myoblast fusion. Moreover, our recently published (Ansseau et al 2016) and new preliminary data indicate that DUX proteins are associated to specific IGF2BP1-dependent ribonucleoparticles (mRNP). In these granules, IGF2BP1 is associated a.o. with ACTB mRNA and regulates the synthesis of non-muscle cytoskeletal actins that is essential in the first steps of myotube formation (elongation, fusion, nuclear dynamics). Later these actins will be replaced by the muscle forms in myofibrils. The DUX4 and DUX4c inductions observed in FSHD muscle cells could therefore interfere with this precise dynamics and contribute to the muscle pathology. Moreover, as DUX4c is normally expressed in almost all myoblasts and as many identified partners are identical for DUX4 and DUX4c, the pathological increase of DUX4/DUX4c proteins in FSHD muscle cells could titrate out some partners and interfere with the normal DUX4c function in muscle and would contribute to explain why this tissue is particularly sensitive to pathological DUX4 expression (one of the FSH Society research priorities for 2016). In this project which is with the follow up of our on-going FSH Society grant (see Annex for a progress report) we therefore want to define the DUX4/4c functions in the cytoplasm with the following specific aims : (1) to monitor DUX4/4c trafficking, cytoskeletal dynamics and nuclear movements during muscle cell differentiation in culture, (2) to identify the RNAs associated with DUX4/4c (3) to study the impact of DUX4/4c gain or loss of function on IGF2BP1-associated mRNP granules (4) to determine the pathological interactions caused by excess DUX4 or DUX4c in muscle cell cultures and in a FSHD mouse model in collaboration with the group of Yi-Wen Chen (George Washington University). The identification of the DUX4/4c peptidic domains involved in the interactions with partners will later be used to select either peptides (using a phage display library), or aptamers that suppress or decrease the interaction with partners involved in toxic pathways. The biological significance of the DUX4/4c-partner interaction will then be evaluated by introducing these agents in FSHD muscle cell cultures and analysing the resulting phenotype. This approach might present a therapeutic interest in FSHD by blocking some DUX4/DUX4c toxic functions. We expect this project will help (i) to define new functions for DUX4 and the poorly studied DUX4c, (ii) to discover their putative interactions through shared partners (iii) to bring new light on the mechanisms of DUX4 toxicity in FSHD muscle and (iv) to propose new therapeutic strategies preventing protein/protein or protein/RNA interactions.
Specific Aims: Facioscapulohumeral Muscular Dystrophy (FSHD) is a muscle wasting disease caused by a genetic mutation in the subtelomeric region of chromosome 4. Healthy individuals have 11-100 repeated D4Z4 microsatellite units, whereas individuals with FSHD have 1-10 D4Z4 units. This genetic contraction makes the 4q35 chromosomal region more accessible to transcriptional machinery and permits transcription of nearby genes including DUX4, the proposed pathological agent in FSHD. DUX4 is a transcription factor that when aberrantly expressed induces transcription of many genes implicated in muscle development, the actin cytoskeleton, regulation, apoptotic signaling pathways, and germline development. While many potential biomarkers of FSHD, including TRIM43, MBD3L2, ZSCAN4, and PITX1, all of which are downstream of DUX4, have been identified from studies of muscle biopsy and of muscle cells in vitro, the pathophysiological role of DUX4 and these target genes has yet to be studied in a humanized in vivo model. The D4Z4 contraction is not replicable in animal models, nor do the effects of DUX4 expression in murine muscle cells replicate those seen in human cells. To overcome these problems, the laboratory has developed a method of xenografting human derived muscle precursor cells, isolated from patients with FSHD and healthy controls, into the tibialis anterior compartment of immune-deficient mice. These cells grow, fuse, and mature into force-producing human muscle fibers within the mouse hind limb. These human muscle fibers can be studied in intact grafts or following dissociation and isolation in culture. This novel in vivo FSHD model will be used to study DUX4 and its potential downstream gene targets, TRIM43, MBD3L2, ZSCN4, and PITX1. The ultimate goal is to determine how their patterns of expression and localization in relation to DUX4 within individual fibers lead to dystrophy – experiments that have never been performed in an FSHD model system before. Further analyses will attempt to reveal the primary defect in FSHD muscle, leading to the creation of targeted FSHD therapies. This study hypothesizes that DUX4 expression in a small percentage of the nuclei of mature human muscle fibers is responsible for activating DUX4 gene targets in the same or nearby myonuclei and that together these induce local muscle wasting, leading to the phenotype seen in FSHD. The hypothesis will be tested by i) comparing the molecular phenotype of intact muscle as well as fibers isolated and cultured from FSHD-derived xenografts to controls and by ii) examining the functional phenotype of FSHD derived xenografts compared to controls. Molecular methods such as qRT-PCR, immunofluorescent labeling, and confocal microscopy combined with electrophysiology-based functional measures will be employed to explore the phenotype in living muscle from FSHD xenografts. This study overcomes the limitations of current models of FSHD associated with viral vectors or incomplete development in culture, as it examines the pathologic consequences of endogenous DUX4 expression and its downstream targets in mature human muscle fibers grown within the mouse hind limb.
Specific Aims: My doctoral and postdoctoral studies aimed at investigating the implication of non-coding RNAs in the regulation of gene expression in bacteria. Therefore, I am quite familiar with gene expression processes as well as their implication in the control of cell homeostasis. One of my objectives for joining Dr. Harper’s lab is my interest in understanding dysregulation processes of gene expression in human diseases (e.g., FSHD). In fact, FSHD arises from complicated pathogenic mechanisms, with epigenetic origins, that ultimately cause overexpression of the transcription factor DUX4, which is toxic to muscle and numerous non-muscle cell types. FSHD presentation is non-uniform, and there may be extreme variability in severity of symptoms, rate of progression and age at onset, even in families with several affected relatives. Similarly, asymmetrical weakness is common. It has been hypothesized that this non-uniformity of presentation might be due to the regulation of DUX4 expression that becomes initiated upon the presence of various but yet undetermined factors. So far, the regulation of DUX4 gene expression is still relatively unexplored, and except SMCHD1, genes that directly modify DUX4 expression have not been identified. We think that DUX4 gene expression modifiers might influence DUX4 toxicity and FSHD disease penetrance. In this proposal, our central hypothesis is that endogenous DUX4-targeted microRNAs (miRNAs) are modifiers of DUX4 expression and toxicity. Specific Aim 1: To define miR-675 regulation of DUX4 gene expression. Specific Aim 2: To define the DUX4-targeted miRNome among all human microRNAs.
The overall, long term aim of this application is to expedite the development of new therapies for FSHD by developing a core FSHD Clinical Trial Research Network (CTRN) composed of 4 institutions (University of Rochester; Kennedy Krieger Institute, Baltimore; Ohio State University, Columbus; and the University of Kansas, Kansas City) with established expertise in FSHD and neuromuscular clinical trials. The specific aims are follows:
Aim 1: Establish an FSHD CTRN infrastructure. To this end, we will:
- a. Develop standard operating procedures (SOPs) and a governance document for the Network
- b. Establish reliance agreements between the IRBs in the CTRN using the existing CTSA IRB reliance model
- c. Institute a data sharing agreement between the network sites.
- d. Create common data dictionary for a RedCap database at the University of Rochester, which can be rolled out at multiple sites, to allow for distributed, online data entry of anonymized data from all four sites.
- e. Create a manual of operations and standard regulatory language for a core set of outcome measures, to improve the efficiency of IRB regulatory submissions.
- f. We will create standardized protocols and CRFs for important FSHD clinical outcomes – wherever possible we will incorporate established NIH FSHD common data elements
- g. Train site clinical evaluators to insure that outcome measure procedures are performed in an identical manner at all sites. Establish inter rater reliability for each of the outcome measures.
Deliverable: All components of Aim 1 will be drafted in CTRN Year 1, then completed by the end CTRN Year 2
Aim 2: Create a system of committees to ensure engagement of all major stakeholders. To this end we will convene:
- a) Patient Engagement Circles comprised of patients willing to convene focus groups on an as needed basis to address specific aims or difficulties encountered in developing the FSHD CTRN, for example defining what would be clinically meaningful to people with FSHD, addressing concerns related to participating in clinical studies, and issues with recruitment and retention.
- b) A Guidance Committee comprised of investigators, patient advocates, patients and industry. This committee will assist the network in fashioning network wide SOPs, public outreach, and selection of projects to be run on the FSHD CTRN.
- c) Steering committee will be fashioned from site investigators, and an FSH Society representative to assist in the day to day running of CTRN network activities
Deliverable: rosters for each committee will be established and meetings held at network start up and prior to implementing the first project in CTRN Year 1.
Aim 3: Conduct a prospective study of outcome measures with funding from the NIH, MDA, and personal philanthropy: A prospective study of 150 patients followed over 18 months will be needed to definitively validate the outcome measures. We will be applying simultaneously to the NIH in a new request for application announcement (http://grants.nih.gov/grants/guide/pa-files/PAR-16-020.html) designed to fund applications for trial readiness in rare neurologic and neuromuscular diseases. The MDA has implemented a new clinical trial pathway for study funding. We feel that we are well positioned for such an application. Moreover, our chances for funding will be significantly improved if we can show that we already have a core FSHD Clinical Trial Network in place.
Deliverable: Funding will be pursued through the NIH, MDA, and personal philanthropic support with a goal of securing funding for a study by the end of CTRN year 1, first patient entered beginning of CTRN Year2. If funding from these sources is limited than the scope of our prospective study will have to be curtailed as a consequence.
2015 grant awards
Summary: Development of FSHD has been linked to the de-repression of the DUX4 gene in the skeletal muscle of affected individuals. However, individuals have been identified who express DUX4 in muscle biopsies, but who do not manifest any clinical signs of the disease. Thus, while derepression of DUX4 may be necessary for the FSHD phenotype, it is not sufficient. This suggests that there may be other factors that modify the FSHD disease phenotype. We used a proteomic approach to screen for DUX4-interacting proteins that may act as disease modifiers and identified the multifunctional C1QBP as one such candidate. C1QBP is known to regulate several of the molecular pathways that are affected by DUX4 expression, including gene expression, oxidative stress, and apoptosis. In our preliminary results we have found C1QBP is dynamically regulated in myogenic cells. It is localized primarily to ribbon-like structures outside of the nucleus in myoblasts, but appears to relocate to the nuclear periphery when they are allowed to fuse into myotubes. Expression of DUX4 leads to increased C1QBP concentration in the nucleus, supporting the hypothesis that DUX4 and C1QBP form functional interactions. Importantly, C1QBP is known to bind to the intra- and extra- cellular signaling molecule hyaluronic acid (HA), which can regulate its phosphorylation state. We have found that that decreasing intracellular HA by treating cells with 4- methylumbelliferone (4MU), an inhibitor of HA synthesis, results in a sharp decline in DUX4-target gene expression. This raises the possibility that it could serve as an FSHD therapeutic. 4MU is of particular interest for development as a drug because it is already being used in Europe to treat biliary dyskinesia, and has had its short-term safety established in several studies. It is also being investigated in both cell culture and animal models as a treatment for specific cancers.
In this project we will evaluate the ability of 4MU to serve as an FSHD therapeutic and investigate its mechanism of action. We have observed that 4MU inhibits the expression of DUX4-target genes, both in myotubes and when DUX4 is overexpressed in myoblasts. We have hypothesized that this is a result of the loss of HA altering the post- translational modifications of C1QBP, which would interfere with its ability to interact with DUX4 and act as a transcriptional cofactor. This could take the form of preventing C1QBP from binding to DUX4 directly, altering the subcellular localization of C1QBP, or by causing changes in C1QBP stability. We will perform studies to evaluate each of these possibilities. Finally, we will use an FSHD mouse xenograft model, established in our lab, to conduct dose-escalation studies to determine if 4MU treatment can inhibit DUX4-target gene expression in vivo. This will better evaluate 4MU as a potential FSHD therapeutic.
Summary: Antisense oligonucleotides (AOs) are chemically modified single-stranded DNA, RNA or chemical analogue molecules which are able to modulate the expression of a specific targeted gene. The advances in the development of antisense chemistries, in particular phosphorodiamidate morpholino oligomers (PMOs), have led to numerous studies investigating the therapeutic potential of antisense technology. AO-mediated exon skipping is currently one of the most promising therapeutic options for Duchenne muscular dystrophy (DMD). Importantly, BioMarin and Sarepta have announced that U.S. Food and Drug Administration (FDA) has accepted for review the submission of a New Drug Application (NDA) for drisapersen (2’OMePS PRO051) and eteplirsen (PMO AVI-4658) for the treatment of DMD.
The overall objective of our project is to suppress DUX4 expression and develop a therapeutic approach for FSHD based on AOs. We have chosen to target the 3’ key elements of DUX4 mRNA and have already obtained robust results demonstrating the feasibility of such an approach. For the first time, we demonstrated in vitro that targeting a functional PAS can be an efficient therapeutic strategy for a genetic disease. We observed that targeting DUX4 3’key elements leads to an efficient extinction of DUX4, does not redirect polyadenylation and prevents aberrant expression of genes downstream of DUX4.
Our goal is now to (i) improve DUX4 mRNA extinction by developing sequence optimized AONs and to (ii) validate these AOs in vivo. Int the first aim, we will otpimize the sequence and chemistry of AO drugs targeting the 3’ key elements of DUX4 mRNA. In the second aim, we will test the body-wide administration of the most active anti-FSHD AO drugs for delivery and effectiveness in animal models after the creation of a new mouse model carrying a reporter gene (LacZ) with the 3’UTR of DUX4 mRNA. Two strategies will be developed: the direct injection of vivo-PMO and the vectorization of the PMO in an AAV vector. In the first case, mice will be subjected to treatment regimens of intravenous systemic delivery of therapeutic optimised AOs in naked form or conjugated to cell-penetrating moieties (eg octaguanidine or CPPs). In the second case, AOs will be vectorized under the control of the U7 promoter as it has been done for exon skipping for DMD for instance. AAVs are now well known to be able to target the muscles in a whole body without toxic effects.
Specific Aims: Autosomal dominant Facioscapulohumeral muscular dystrophy (FSHD) is the third most prevalent muscular dystrophy, affecting 1 in 20,000 individuals. FSHD was formally classified as a major form of muscular dystrophy in 1954, but the pathogenic events leading to the disease have only recently started coming into focus. Several studies now support an FSHD pathogenesis model involving aberrant expression of the primate specific DUX4 gene, which encodes a myotoxic transcription factor. The emergence of DUX4 represented a momentum shift in the FSHD field as it provided an important target for therapy design. Indeed, as FSHD is currently untreatable, developing effective therapies is a critical need in the field. An FSHD treatment should center on inhibiting DUX4, which could be accomplished by silencing the gene or transcript, or negating the toxic effects of the DUX4 protein. The overall objective of this study is to identify, characterize, and ultimately inhibit DUX4 protein modifications that may contribute to its toxic properties in FSHD muscle. Delineating how DUX4 protein function is regulated is an important unmet need in the field.
The DUX4 gene encodes a transcription factor that activates downstream toxic pathways, including apoptotic cascades. I hypothesized that post translational modification (PTM) may be one important mechanism affecting DUX4 protein function. PTMs play key roles in ligand binding affinity, subcellular localization and protein stability. My primary goal was to first identify whether DUX4 could be post translationally modified, then subsequently map DUX4 PTMs and determine their contribution to DUX4-induced toxicity. My aims are designed to define the role of DUX4 PTMs, which will allow us to potentially understand its protein function and regulation. By accomplishing this goal, we hope to establish a framework for therapeutic intervention designed to disrupt DUX4 modifications and prevent myotoxicity.
Aim 1: To define DUX4 post-translational modifications The DUX4 transcription factor is associated with muscle pathology in FSHD and is toxic to numerous other non-muscle cell types. However, some cells and tissues seem to resist DUX4-associated damage, including the testes, where DUX4 is naturally expressed at high levels, as well as muscles of non-manifesting FSHD carriers. The mechanisms by which some cells resist DUX4-associated damage are unknown, but it is likely that DUX4-modifier genes may impact disease penetrance. Since PTMs can profoundly influence transcription factor activity, I hypothesized that the DUX4 protein may also be regulated by PTMs, and the enzymes that mediate these PTMs could therefore impact DUX4 toxicity. In preliminary studies, I found that the DUX4 protein is modified by methylation and phosphorylation using mass spectrometric analysis of DUX4. In this aim, I will define the PTM signature of DUX4 in numerous cell types, including human myoblasts and primate testes, which endogenously express DUX4. Differences in the modification signature of DUX4 isolated from these cell types will provide insight about tissue-specific regulation of DUX4 protein, and may provide information about the differential toxicity of DUX4 in tissues or cells.
Aim 2: To examine the role of phosphorylation on DUX4 function My preliminary results revealed phosphorylation of numerous DUX4 residues in both the N and C-terminal domains. In this aim, I propose to examine the effects of each phosphorylation event using mutagenesis to irreversibly mimic or ablate DUX4 phosphorylated residues. I will then determine the effects of these DUX4 mutants in vitro using several outcome measures that I have previously established in my preliminary studies. These include DUX4 DNA binding affinity, assessment of the impacts on key ligand interactions, DUX4 dimerization, cellular toxicity and gene target activation. This work will help establish an important first step toward developing therapies that could prevent DUX4-mediated toxicity, by differentially affecting the phosphorylation status of DUX4.
Summary: We will submit formal grant applications to foundations, including MDA and the FSH Society, and are considering seeking some industry funding. However, this will take time (several months), and we no longer have discretionary funds to support the mouse colony. We want to expand, characterize, and publish this model as soon as possible, and we are seeking bridge funding for this purpose. It is our goal and priority to make this model available to anyone in the field who wants it, as soon as is practicable.
Summary: Our laboratory, together with A. Belayew’s laboratory, originally proposed that aberrant expression of DUX4 is harmful to cells, contributing to the pathogenesis of FSHD. We demonstrated that DUX4 is a nuclear protein, endogenously expressed in cultured FSHD myoblasts, pro-apoptotic and cytotoxic when expressed in transfected cells. We recently analyzed the DUX4 molecular domains contributing to its toxicity, subcellular transit and nuclear location. In these studies we recognized an LLXXL motif at the C-terminal region of DUX4, which is present in co-regulators of nuclear hormone receptors (NRs). Preliminary studies from our laboratory showed that DUX4 is a co-regulator of the progesterone NR. We also found that progesterone protects cultured cells from the toxic effect of DUX4. In this project we will study the role of DUX4 as a co-regulator of NRs of sex hormones as well as the protective effect of sex hormones on the toxicity of DUX4. These studies are relevant to the understanding of the normal function of DUX4 as well as its pathogenic role in FSHD and the future rational approaches for the treatment of FSHD patients.
Summary: Facioscapulohumeral dystrophy (FSHD) is a common but unique form of muscular dystrophy requiring multiple factors to create a ‘permissive’ state for disease manifestation. Over recent years, several genetic (DUX4) and epigenetic (hypo-methylation) factors have been linked to FSHD pathogenesis; however, it has become clear that the field has not elucidated all factors required for disease manifestation. Mounting clinical evidence suggests the existence of modifier genes with the capacity to regulate DUX4 transcript and/or protein function. Recent advances in genome-editing technologies proposed for use in this project now should enable us to uncover these remaining missing links. Through the systematic introduction of loss-of-function mutations into genomic DNA, we can interrogate the genome for answers that may explain the phenotypic variability between patients, as well as the non-penetrant effects of DUX4 in some individuals. In this project, we propose a targeted genome-scale knock-out screen to identify genes that can reduce the phenotypic impact of DUX4 expression when inactivated. We hypothesize that there exists gene targets of DUX4 whose loss will render DUX4 unable to trigger a dysregulated cascade of gene expression, thus abrogating its toxicity. These candidates likely serve as genetic modifiers of FSHD, and will be readily identified by downstream sequencing and computational analysis for detection of CRISPR target genes enriched within these DUX4 ‘resistant’ cell populations. This will allow the generation of a complete list of gene candidates with the potential to influence the pathogenic outcomes associated with DUX4 misexpression. Identified gene hits will be cross-referenced to our whole-genome sequencing data of nonmanifesting carriers to search for sequence variants that may enable us to narrow down promising candidates for functional follow up studies. Validation of candidate modifier genes will be performed in our established zebrafish model of FSHD for rescue of phenotype to confirm functional significance. Additionally, we will revert to our repository of FSHD patient cells to genome edit our candidate genes under these permissive allelic conditions, and subsequently measure changes in known FSHD biomarker expression. FSHD is a challenging disease whose remaining unanswered questions cannot be accomplished alone. Hence, our proposal involves a multi-institute collaboration, bringing together a wealth of patient resources (Wellstone Center), the latest in genomic technology (Broad Institute), and a well-established animal model of FSHD (Boston Children’s Hospital). Not only will the identification of these modifier genes for DUX4 resistance provide valuable insights into FSHD disease pathogenesis, but they will also present as solid leads that can be directly targeted for therapeutic intervention in humans with FSHD.
The long term goal of this research project is to establish a quantitative assessment tool to evaluate changes in mobility status of persons with FSHD. We will use a portable wireless motion analysis system to instrument a timed up and go, postural sway during quiet standing, and arm range of motion. We plan to build on an existing University of Kansas Medical Center Frontier’s pilot grant which will identify the specific outcome metrics obtained with portable wireless motion sensors which are important for examination in persons with FSHD and determine the reliability and cross sectional validity of those metrics. In the present FSHD Society study, we propose to extend our current pilot study to add 6 and 12 month follow up visits. We will conduct a 12 month longitudinal study in 20 genetically confirmed and clinically affected FSHD participants (10 mild to moderately affected, and 10 moderate to severely affected) to determine the responsiveness of wireless motion analysis to disease progression in FSHD, determine the minimal detectible change and minimally clinically important change in these metrics, and use factor analysis to create summary scores (e.g. upper extremity, lower extremity) for future clinical trials.
Summary: Funds are being requested from the FSH Society to cover the remaining months of graduate student Yuanfan “Tracy” Zhang in the Wagner lab. Tracy is a 5th year Cellular and Molecular Medicine graduate student who works exclusively on FSHD. Her thesis work is to establish, validate and use a novel model of FSHD. She established and validated the human skeletal muscle xenograft for FSHD which she published as a first author in Human Molecular Genetics (Zhang et al., Hum Mol Gen 2014, 23: 3180-3188). She is now using the model to show proof-of-concept of antisense oligonucleotide knockdown of DUX4-fl in FSHD. While this work is generally supported by the FSHD Wellstone at UMMS, Tracy is no longer supported by the Wellstone and funds are being requested to cover her salary and benefits to finish this project.
PROJECT SUMMARY: The goals of our studies are to identify pathogenic mechanisms and develop new therapeutic strategies for facioscapulohumeral muscular dystrophy (FSHD). We discovered that full-length isoform of double homeobox protein DUX4 (DUX4-FL), but not DUX4-S (short isoform of DUX4), inhibits protein turnover and leads to abnormal ubiquitin expression and nuclear aggregation of TDP-43, one of the aggregation-prone and an RNA/DNA-binding proteins previously associated with amyotrophic lateral sclerosis (ALS) and inclusion body myositis (IBM)(Homma et al., 2015). These phenotypes were not side effects of DUX4-FL-induced cell death and were enhanced when cell death was blocked by caspase inhibitors. These data suggest that DUX4-FL induces abnormal protein degradation, which can in turn lead to cytotoxicity. Importantly, the abnormal deposition of ubiquitinated protein and nuclear aggregation of TDP-43 were observed in DUX4-FL-expressing cells from its endogenous promoter as well as when exogenously expressed. Thus, DUX4-FL produced from its endogenous promoter is sufficient to promote pathogenesis, and our results identify inhibition of protein turnover and impaired proteostasis as potential pathological mechanisms in FSHD. We now propose to identify mechanisms that underlie the DUX4-FL-induced inhibition of protein turnover and promotion of abnormal protein aggregation. Under Specific Aim 1, we will identify the mechanisms by which DUX4-FL expression inhibits proteasome function. We will isolate proteasomes from DUX4- positive and -negative myotubes and the amount and activity of proteasome will be examined. If intrinsic proteasome activity and/or the amount are unchanged by DUX4-FL expression, then we will investigate indirect mechanisms by which ubiquitinated proteins could abnormally accumulate in DUX4-FL-positive myotubes. Under Specific Aim 2, we will examine FSHD muscle biopsies to identify if there are signs of dysfunction of protein degradation system. We will immunostain FSHD and control tissues with antibodies for ubiquitin, TDP-43 and other proteins that are associated with myopathies and protein aggregation diseases. The outcome of this study could use as clinical marker(s) for FSHD. Thus our proposed studies will provide valuable insights into the mechanisms of DUX4-FL induced pathology in FSHD that might share with some other myopathies and/or protein aggregation diseases. This new knowledge could develop potential new therapeutic strategies based on regulating proteasome activities and identify new clinical biomarker(s) for FSHD.
Aim 1. Doxycycline inducible lentivirus-SMCHD1 in FSHD1 and FSHD2 muscle cells. We will determine the effectiveness of increased SMCHD1 expression to suppress DUX4 in FSHD with different mutations using doxycycline inducible lentivirus-SMCHD1 (a and b). SMCHD1 is composed of exons 48 and has ATPase and Hinge domain. We will determine the critical region of SMCHD1 to suppress abnormal DUX4 in FSHD muscle cells using doxycycline inducible lentivirus-SMCHD1 that has short SMCHD1 coding sequence (c). a. Ability to suppress DUX4 in FSHD1 with i) Slightly lower than normal repeat size (7-10 unit) ii) Severe contraction (1-6 unit) b. Ability to suppress DUX4 in FSHD2 with i) Haploinsufficient mutation ii) Dominant negative mutation c. Ability to suppress DUX4 in a range of SMCHD1 coding sequence in FSHD muscle cells. i) Full length (exons 1-48) ii) Short length (exons 1-10, 1-20, 1-30, 1-40)
Aim 2. rAAV6-SMCHD1 in FSHD muscle cells and D4Z4-2.5 mice. We will determine the effectiveness of increased SMCHD1 expression to suppress DUX4 with recombinant adenoassociated virus 6 (rAAV6) – cytomegalovirus (CMV) – SMCHD1 in FSHD1 and FSHD2 muscle cells and D4Z4-2.5 mice (FSHD1 model mice). In addition, to distinguish the effect of SMCHD1 in whole body from muscle on suppression of DUX4 in FSHD, we will choose CMV promoter and enhancer/promoter regions of muscle creatine kinase and α-myosin heavy-chain (MHCK) genes and administrate rAAV6-CMV-SMCHD1 and rAAV6-MHCK-SMCHD1 into D4Z4-2.5 mice. a. Ability to suppress DUX4 in FSHD1 and FSHD2 muscle cells using rAAV6-CMV-SMCHD1. b. Ability to suppress DUX4 in D4Z4-2.5 mice using rAAV6-CMV-SMCHD1 and rAAV6-MHCK- SMCHD1.
In this application, Aim 1 will be the proof of principle experiments showing that higher SMCHD1 will be effective as a potential therapy and Aim 2 will develop a method for delivery that might eventually be suitable for pre-clinical or clinical studies based on rAAV6.
PROJECT SUMMARY: The Double homeobox (DUX) genes map in 3.3-kb repeated elements that constitute a family with hundreds of members dispersed into the human genome. Even if the evolutionary conservation of their sequences argues in favor of a functionality, they were long considered as pseudogenes and thus poorly studied. However, several DUX genes are expressed in healthy muscle cells. Our group has characterized the DUX4 gene that causes FSHD and the homologous DUX4c gene (both located at 4q35). Both encoded proteins are highly similar transcription factors and only differ in the carboxyl-terminal region. There is no known orthologue of these genes in rodent. However, mouse Duxbl, a paralogue sharing similarities with DUX4 and DUX4c, was previously shown to play roles during myogenesis. DUX4c is expressed in healthy muscle and induced in FSHD and Duchenne muscular dystrophy (DMD). Our previous data suggested a role for DUX4c in normal human muscle regeneration and its activation (as in FSHD) could impact muscle regeneration in several myopathies. Deciphering the function of unstudied human muscle proteins should increase our understanding of physiological and pathological mechanisms of the skeletal muscle. The present project stems from our identification of putative and validated protein partners that hint to unexpected cytoplasmic functions for the DUX4/DUX4c proteins in myofibril organization and mRNA translation control. Moreover, in addition to the endogenous DUX4c nuclear localization in myoblasts, we found a cytoplasmic localization of this protein in differentiating myoblast at particular times. We also observed DUX4 cytoplasmic translocation during myoblast differentiation following its overexpression. As a lot of identified partners are identical for DUX4 and DUX4c, the pathological increase of DUX4/DUX4c proteins in FSHD muscle cells could titrate out some partners and interfere with the normal DUX4c function in muscle and would contribute to explain why this tissue is particularly sensitive to pathological DUX4 expression (one of the FSH Society research priorities for 2015). To further investigate these observations, we would (1) monitor DUX4/DUX4c protein trafficking in live muscle cells (time-lapse microscope) by using fluorescent ligands to label DUX4/DUX4c expressed as HaloTag fusion proteins and by using different nuclear export/import inhibitors; (2) produce additional antibodies specifically targeting DUX4c (3) validate DUX4/DUX4c interactions with partners that play major roles in myofibril organization or mRNA translation using in situ proximal ligation assays; (4) compare the interactions of selected partners and DUX4/4c in healthy and FSHD differentiating myoblasts (5) map the specific DUX4/DUX4c peptidic domains that interact with validated partners using DUX4/DUX4c specific or deletion mutants and HaloTag pull down; The latter will be used to screen a phage display library with the mapped interaction domains to select peptides that suppress or decrease the interaction with DUX4/DUX4c of partners involved in toxic pathways. The biological significance of the DUX-partner interaction will then be evaluated by introducing the blocking peptide in FSHD muscle cell cultures and analysing the resulting phenotype. This approach might present a therapeutic interest in FSHD by blocking DUX4/DUX4c toxic functions. We expect this project will help (i) to define new functions for DUX4 and the poorly studied DUX4c, (ii) to discover their putative interactions through shared partners (iii) to bring new light on the mechanisms of DUX4 toxicity in FSHD muscle and (iv) to propose new therapeutic strategies preventing protein/protein interactions.
PROJECT SUMMARY: In response to a request from the FSH Society, NDRI proposes to develop and implement a resource to recover surgical and post mortem human bio-specimens and distribute them to approved investigators. This resource will utilize NDRI’s experience, expertise and established systems to expand and enhance the type, number and quality of human tissues available to the FSH research community. lt is proposed that NDRI’s Private Donor Program will collaborate with FSH to recover and distribute tissues from patients who participate in the FSH Registry and who have provided consent for the recovery of tissues and organs for research. In addition to providing all resources required to recover tissues post mortem and from surgical procedures, NDRI will provide informational materials to the FSH Society for distribution to potential registry participants, as well as IRB-approved templates for obtaining informed consent from patients and authorization to donate from family decision makers.
Sequence-specific nucleases cut the DNA molecule at defined sequences. If the sequence-specific nuclease is designed to a particular site, for example the FSHD locus, they allow scientists to make designed changes at that site. The Kyba Laboratory has been using working with two types of sequence-specific nucleases, called a zinc finger-nuclease, and TALENs, however a new technology has recently come on the scene, referred to as CRISPR/Cas9. This technology allows much more rapid development and testing of genome editing approaches. This grant allows the lab to apply this new technology to genome editing projects. The grant has 3 overall goals: 1. They specifically plan to use CRISPR/Cas9 to modify the “pathogenic poly A sequence”, which is the FSHD-causing sequence that lies on the far side of the D4Z4 repeats. The cells they will use for these studies are human induced pluripotent cells from FSHD donors. These are cells from an adult donor that have been reprogrammed to behave like embryonic cells, with unlimited proliferation and differentiation potential, thus once genetically corrected they should be able to generate muscle stem cells that lack the FSHD mutation. 2. The funding will also allow the lab to perform genome editing on embryonic stem cells carrying the FSHD mutation. 3. The lab will use the technology to make defined changes in the way that DNA at the FSHD locus on chromosome 4 is packaged, in order to test models for the molecular mechanism underlying FSHD. These studies are proceeding apace and promise to shed light on new approaches for therapy development. For further details see awards from August 2013.
2014 grant awards
Abstract: The pathogenesis of facioscapulohumeral muscular dystrophy (FSHD) pathogenesis is complex and not yet fully understood. Recent detailed genetic studies have significantly increased our knowledge of this enigmatic and multifaceted disorder, suggesting the aberrant genetic events during very early myogenesis. The establishment of human induced pluripotent stem cells (hiPSCs) ushered a new era in biomedicine and provide unprecedented opportunities for modeling human genetic diseases. The Lee lab has developed a novel strategy to direct the hiPSCs into myoblasts as well as gene-targeting approach with CRISPR/Cas9 system. Here we propose to isolate pluripotent cells, somite cells and myoblast cells of FSHD-hiPSCs using the established techniques, followed by detailed transcriptional analysis. Our proposed studies will shed light on the FSHD pathogenesis in stage-specific manner during very early human myogenesis.
Abstract: FSHD is linked to chromosomal 4q35 region, that contains a D4Z4 array of up to 200 units. The most common form, autosomal dominant FSHD1, is caused by a contraction of the 4q D4Z4 array to less than 11 units, whereas FSHD2 is caused by reduced levels of functional SMCHD1 protein (Structural maintenance of chromosomes flexible hinge domain-containing 1). Although with different mechanisms, both genetic defects lead to DNA hypomethylation at D4Z4 on 4qter causing chromatin relaxation. This genomic modulation provides a transcriptionally permissive chromatin environment that is associated with the expression of DUX4, the best candidate FSHD gene, enclosed within each D4Z4 unit. DUX4 expression requires also the presence of a polyadenylation signal (PAS) distal to the last D4Z4 unit, which stabilizes DUX transcript. There are two different allelic forms of the region distal to the D4Z4 array, A and B. Although a D4Z4 array followed by an A “Telomere” is also present on 10q, a functional PAS sequence has been identified almost exclusively on 4qA alleles. Ultimately, vast majority of FSHD1 and FSHD2 subjects show hypomethylation at D4Z4 region followed by an A allele containing a functional PAS. Currently, FSHD diagnosis is based on the identification of shortened 4q arrays (FSHD1) or the presence of mutations in SMCHD1 (in FSHD2) and the assessment of the A/B genotype. In addition, methylation analysis of the proximal D4Z4 units is performed, using methylation sensitive restriction enzymes or by bisulfite sequencing of the overall D4Z4 units. Although hypomethylation is significantly associated with FSHD1 and FSHD2, it is not diagnostic per se because of the lack of information about the presence of permissive alleles (alleles that contains a polyadenylation signal – PAS) and of the interference of not-pathogenic arrays. For this reason, the diagnostic flowchart for FSHD considers hypomethylation as a secondary step to distinguish FSHD1 from FSHD2. Our project aims to the introduction of a new assay that combines the different key features found in FSHD subjects. We propose methylation analysis of 10 CpGs within the 3’ portion of the distal DUX4 copy (DUX4-fl), that is specifically expressed in muscles of FSHD patients. Despite the low complexity and the presence of repetitive elements in the region (pLAM), we were able to design PCR assays, on bisulfite treated DNA, that are specific for the presence of PAS sequence in the A allele. Preliminary results in a subset of FSHD1, FSHD2 and Control subjects showed highly significant differences of methylation levels between affected and unaffected subjects in 8 out of 10 CpGs tested, strongly supporting the potential usefulness of this assay for FSHD diagnosis. Here, we propose:
- To develop additional assays to quantify the number of permissive alleles in order to assess whether different allelic combinations are relevant in the identification of diagnostic threshold;
- To analyze a large cohort of well genotyped FSHD patients and normal controls for precise evaluation of methylation threshold between affected and unaffected subjects;
- To assess specificity of this assay for FSHD disease by testing peripheral blood leukocytes DNA (PBLs) from individuals with unrelated muscular dystrophies;
- To analyze the prognostic potential of this assay by correlating methylation levels with different clinical severity scores;
- To study possible methylation differences distal to the D4Z4 array, between PBLs and muscle biopsies.
Abstract: The pathophysiology of Facioscapulohumeral Dystrophy (FSHD) is poorly understood and understudied. This FSH Society fellowship award to Dr. Jun Udaka will support comprehensive and detailed physiological investigations of force generation and calcium signaling in skinned and permeabilized single fiber preparations ofFSHD and unaffected control muscle biopsies. Proposed studies address whether FSHD muscles are defective in: 1) troponin-mediated Ca2+ signaling for myofibrillar force generation, 2) sarcoplasmic reticulum (SR) calcium release, 3) myosin ATPase activity, and/or 4), contraction fatigue. Additional studies investigate the roles oftitin and extracellular matrix (ECM) in muscle fiber elasticity as contributing factors in FSHD disease pathology. Findings wiII identify muscle proteins and contractile processes that become dysfunctional during FSHD disease progression and reveal the underlying pathophysiology of FSHD muscle weakness to enable the development and evaluation of FSHD therapeutics in pre-clinical and clinical studies.
Abstract: Facioscapulohumeral muscular dystrophy (FSHD) is a disease group which can be subdivided into two groups (FSHD1, OMIM# 158900 and FSHD2, OMIM# 158901). All patients have in common a muscular dystrophy affecting the facial and the upper limb muscles. In both groups, additional features are known such as hearing loss and mild to moderate eye abnormalities. FSHD1 is inherited in an autosomal dominant fashion involving D4Z4 repeat on chromosome 4q35 [1]. The inheritance of FSHD2 is more complex following a digenetic model which includes heterozygous mutations in SMCHD1 which modulate the severity and an haplotype on chromosome 4 which is permissive for DUX4 [2]. Some years ago, a consanguineous family from Italy suffering from an FSHD-like phenotype came to our attention. Both parents were unaffected whereas all four children show a progressive phenotype. The patients show facial and upper limb muscular weakness which became more severe with age. A muscle biopsy from one proband was investigated and showed mild fibrosis, targetoid fibers and an neurogenic component with increased abundancy of type one muscle fibers (JJ Martin, Antwerp). Beside muscular features, all affected individuals reported on eye abnormalities such as myopia in early infancy. A detailed investigation of the eye and the eye fundus revealed an atrophy of the optic nerve in all affected individuals, determining progressive blindness. Furthermore, deep investigation of multiple serum and urine parameters showed an increased level of 3-methylglutaconic acid in the urine and in the serum samples analyzed. Additionally, creatine kinase values are also increase in all individuals tested. The affected individuals from this exceptional family suffer from a disease which combines features known for FSHD and optic nerve atrophy. Furthermore, metabolic alterations which might point to mitochondrial dysfunction were identified which could lead to a better understanding of the affected gene product. Since unaffected parents were second-cousins, recurrence in sibship strongly suggested autosomal recessive inheritance. So far, using various molecular genetic techniques we were not able to detect the causative genetic defect in our index family. Due to the fact that no deletions and causative coding mutations as well as alterations in regulatory regions around CCDC67 could be identified, it is very likely that a non-coding mutation is the cause of this disease. To identify the causative alteration ,whole genome sequencing (WGS) is the method of choice. Rawdata analysis and interpretation may be conducted in the Instute of Human Genetics at Charité (University of Berlin) where both the applicant fellow and his supervisor have acquired an international expertise Moreover, they collaborate tightly with strong bioinformatics group which is experienced in evaluation of large scale genetic data [3, 4]. We are planning to analyze whether larger genomic rearrangements such as an inversion or translocations are present which could explain for example the misexpression of CCDC67. Furthermore, mutations in other intergenic and potential regulatory structures will be investigated. After the identification of the causative mutation, we plan to perform functional investigations in in vitro and in vivo models. We are experienced in cells culture driven investigations of genetic disease [5] and also the generation of mouse models using for example CRISPR/Cas is established in our lab and in the collaboration group at the Max-Planck Institute of Molecular genetics.
Abstract: FSHD is a hereditary human muscular dystrophy affecting groups of muscles in the face and shoulder, characterized by the asymmetry of these muscle symptoms, and additional non-muscular symptoms including hearing loss and retinal vascular abnormalities. FSHD is caused in most cases by chromosomal abnormalities at 4q35, leading to ex cess production of a transcription factor, DUX4, thereby triggering a cascade of gene de-regulations. However, although necessary, DUX4 activation is not sufficient to trigger the symptoms on its own, implying the existence of disease modifiers necessary for the symptoms to appear. My team studies neuromuscular development and the pathologies resulting from alterations of these processes. We have recently started to work on Facioscapulohumeral muscular Dystrophy (FSHD), a hereditary human myopathy characterized by degeneration of muscles in the face and shoulder area, after having found that disruption of the Fat1 cadherin gene in mice caused muscular and non-muscular symptoms resembling those of FSHD, and shown that alterations of the FAT1 locus in humans, located near the FSHD critical region on chromosome 4q35, were associated with FSHD, identifying FAT1 as a modifier gene in FSHD and as a key player of muscular pathologies ([1] : Caruso, PLoS Genetics, 2013). Fat1 ablation in mice causes abnormalities in shape of selective groups of muscles and leads to regionalized muscle wasting at postnatal stages, the map of affected muscles being highly similar to the map of muscles affected in FSHD [1]. Such a possibility was investigated in collaboration with Pr. N. Levy and M. Bartoli, La Timone, Marseille, and J. Dumonceaux, Myology Institute, Paris. a) We found reduced FAT1 expression levels in muscles of fetal [1], but also adult ([2]: Mariot et al. submitted) FSHD cases; b) We identified human mutations in the FAT1 locus segregating with FSHD: i) Heterozygous deletions of a putative regulatory enhancer, predicted to cause tissue-specific depletion of FAT1, co-segregate with FSHD [1]; ii) heterozygous point mutations, either perturbing splicing of FAT1, or leading to deleterious aminoacid changes, were found in FSHD-like patients carrying neither 4q35 alterations nor mutations in SMCHD1 ([3]: Puppo et al., under revision, coll Bartoli/Levy). Thus, FAT1 is a compelling novel FSHD modifier gene, which tissue-specific loss-of-function is sufficient to recapitulate FSHD-like symptoms on its own, and which deregulation was found to co-occur with FSHD. This collaborative work was the objective of a network grant from FSHD Global for which I was the coordinator. In addition, part of this work was also supported by the FSH society through a grant to V. Mariot, working with J. Dumonceaux. My group first initiated work aimed to elucidate in which cell type FAT1 functions were relevant to FSHD-like phenotypes. Through cre/lox-mediated ablation of Fat1 functions in premigratory myoblasts (Pax3-cre), we showed that Fat1 is required in the myogenic lineage to control myoblast migration polarity [1]. We are currently studying the consequences in developing embryos and adult mice of ablating Fat1 in muscle, neurons and mesenchyme. This work has previously received support from the FSH society through a postdoctoral fellowship to Angela Zimmermann in my lab, and is being prepared for publication. The present project aims to extend this work through following approache: We will evaluate the relative frequency of any genetic alteration occurring in the FAT1 locus among classical FSHD1 patients, with particular focus on patients with retinal vascular symptoms, also present in Fat1-deficient mice, to determine whether alteration of FAT1 expression occurs as a result of DUX4 expression, or synergizes with DUX4 expression to cause FSHD symptoms. Results of this project will help understanding to what extent the phenotypes caused by perturbations of FAT1 functions contribute to the appearance of FSHD symptoms, and will be instrumental to elaborate novel therapeutic strategies for FSHD patients.
Facioscapulohumeral muscular dystrophy (FSHD) is a prevalent and currently untreatable myopathy. FSHD is caused by the misexpression of DUX4, a germline transcription factor, in post-mitotic muscle cells where it activates a germline transcription program and also induces expression of retroelements and repetitive sequences. Ectopic expression of DUX4 triggers cell death in a variety of cells including primary myoblasts and immortalized epithelial cells via an unknown mechanism. We recently discovered that DUX4 reduces the efficiency of a cytoprotective, RNA quality control pathway called the nonsense mediated RNA decay (NMD), thus stabilizing hundreds of aberrant RNAs. It is known that reduced NMD efficiency can affect cellular proteostasis due to expression of malfolded proteins, which can in turn lead to cytotoxicity through the unfolded protein response (UPR). Hence we hypothesized that DUX4-induced reduction in NMD efficiency leads to the stable expression and translation of aberrant RNAs, generating toxic proteins that cause cell death, possibly through UPR-mediated apoptosis. In Aim 1, we will identify the mechanism by which DUX4 expression reduces NMD efficiency. In Aim 2, we will determine the contribution of reduced NMD to DUX4-induced cytotoxicity and elucidate the downstream mechanisms responsible for this phenomenon. These studies will provide valuable insights into the mechanism of DUX4-induced cytotoxicity and uncover potential novel avenues for therapeutic intervention for FSHD.
Promoting the appropriate epigenetic repression of DUX4 is a therapeutic strategy for FSHD that addresses the underlying mechanism of disease pathology. However, the molecular details of DUX4 de-repression are not completely understood and few specific targets amenable to small molecule drug intervention have been identified. We have used a chemical genetics approach to identify a key role for the bromodomain and extraterminal domain (BET) proteins in the epigenetic switch that activates DUX4. The experiments proposed here will extend these findings by confirming by genetic means the specific BET family member(s) involved in pathogenic DUX4 expression. This will be accomplished by a combination of RNAi technology and overexpression studies. In addition, we will similarly determine the involvement of mediators of the BET pathway of transcriptional activation including the role of protein acetylation. We will aslo determine the functional effects of BET inhibitors (BETi) on FSHD muscle biology in vitro. A 24 h pulse of BETi results in a sustained decrease in expression of DUX4 and its downstream targets in cultured myotubes without long-term interference with muscle differentiation. These data demonstrate that the pharmacodynamics of DUX4 inhibition and undesirable effects on muscle cells are distinct. We propose to perform a more detailed analysis of the effects of BETi on FSHD myoblasts and myotubes by comprehensive gene expression and functional assays. In addition, we will assess protection of FSHD muscle cells from DUX4-induced apoptosis during myotube differentiation.
This proposal seeks to establish an FSHD Clinical Trials Network composed of academic research centers working collaboratively in developing, testing and validating clinical outcome measures and biomarkers. Creating this network will significantly increase the likelihood that promising therapeutic interventions in FSHD comes to clinical trials and that those trials will have meaningful outcomes.
2013 grant awards
Summary (Provided by Applicant): Transcriptional de-repression of DUX4 gene due to epigenetic changes of the D4Z4 region is believed to cause FSHD. In our preliminary study, we identified that poly (ADP-ribose) polymerase 1 (PARP1) interacted with the promoter of the DUX4 gene. Interestingly, the interaction was only observed in the FSHD myoblasts but not control cells, suggesting that the interaction may be part of the disease mechanism of FSHD. PARP1 is a nuclear protein, which functions in various cellular processes and has been shown to play critical roles in regulating gene expression. Several studies showed that, at the promoter of a target gene, PARP1 binds to DNA methyltransferase 1 (DNMT1) and suppresses its function by poly-ADP ribosylation. As a consequence, expression of the target gene is de-repressed due to hypomethylation of its promoter region. Interestingly we identified that DNMT1 co-localized at the DUX4 promoter region in our preliminary study. In addition, FSHD myoblasts treated with PARP1 inhibitor showed reduced expression of ZSCAN4, a marker of DUX4 expression. Based on current knowledge and our preliminary data, we hypothesized that the interaction among the PARP1, DNMT1 and the DUX4 promoter contributes to the DNA hypomethylation of the region, and may further influence the expression of DUX4 in FSHD myoblasts. The goal of this study is to test one synthetic and one dietary PARP1 inhibitor for their effects on DUX4 expression and further investigate the involvement of PARP1 and DNMT1 in FSHD. In aim 1, we will determine the effects of PARP1 inhibitors on DUX4 expression and cell phenotypes of FSHD myoblasts. In aim 2, we will determine whether DNMT1 directly interacts with the DUX4 promoter region in FSHD myoblasts. In aim 3, we will determine whether PARP1 is a direct regulatory target of DUX4. The findings of the study will provide insights of the involvement of PARP1 in FSHD and have a direct impact on developing therapeutics for FSHD which does not have an effective treatment currently.
Summary (Provided by Applicant): FacioScapuloHumeral Dystrophy (FSHD) is one of the most common myopathies and 2 loci of the disease have been characterized. The first one is located in the subtelomeric region of chromosome 4 and is mutated in 95% of FSHD patients (named FSHD1). This region is composed by a 3.3 kb tandemly repeated sequence named D4Z4. In the general population, the number of repeats varies from 11 to 150, whereas FSHD1 patients carry between 1 and 10 repeats. The second one is located in chromosome 18 and is mutated in 5% of the FSHD patients in whom mutations in the SMCHD1 gene have been found. Despite the different genetic origins of the disease, all patients are phenotypically indistinguishable and share common molecular features, among them the expression of a protein named DUX4. DUX4 is a transcription factor encoding a potential homeobox protein which is highly toxic after overexpression by mis-regulating more than 500 genes. DUX4 ORF is present in each D4Z4 unit but only the most telomeric unit might be able to produce a DUX4 mRNA stabilized by the addition of the poly(A) tail induced by 4qA sequences downstream the D4Z4 array. Because DUX4 is the common pathogenic target between FSHD1 and 2 patients, our goal is to perform gene editing using transcription activator-like effector nuclease (TALEN) and CRISPR/Cas9 technology to modify the FSHD locus and permanently inhibit DUX4 expression. We have chosen to develop 2 strategies: (i) to remove the entire D4Z4 array because individuals with such deletions exist and do not present muscular pathology and (ii) to mutate the DUX4 poly(A) signal since it has been shown that a single point mutation in this poly(A) sequence is sufficient to inhibit DUX4 mRNA expression by modifying its stability. Specific aims will include: (i) designing nucleases with the best activity and sequence-specificity and optimizing the genome engineering strategy (ii) select FSHD cells carrying D4Z4 and 4qA sequence modifications for DUX4 inbhibition (iii) testing the therapeutic benefit of D4Z4 genome engineering in appropriate cell culture and animal models by performing several phenotypic measures to assess the consequences of the targeted mutations of the D4Z4 array on FSHD hallmarks. There are a number of advantages to of our proposed approach over other therapeutic strategies currently under investigation for FSHD. There will be no need for repeated long-term administration of treatment since genome editing offers the possibility of permanent correction following transient nuclease activity for the lifetime of the modified cell and its progeny. The benefit of this as a clinical therapy in terms of cost, toxicological and immunological risk is obvious. Moreover, this approach would be useful for all FSHD cases, whatever the precise mutation/contraction involved.
Summary (Provided by Applicant): DUX4 has been identified as an underlying insult in FSHD, but the mechanisms by which DUX4 contributes to FSHD pathologies is unclear. Our central hypothesis is that the DUX4 transcription factor is involved in protein-protein interactions that influence its ability to induce toxicity in muscle cells and ultimately contribute to FSHD. The two DUX4 N-terminal homeodomains are responsible for its ability to bind specific sequences of DNA. C-terminal residues 160-424 of the DUX4 transactivation domain are essential for inducing toxicity in the muscle, however the mechanisms by which the C-terminal domain mediates DUX4 activity are unknown. We propose that the DUX4 C-terminal domain is involved in the recruitment of proteins that influence its ability to transactivate normal and toxic genes. Our preliminary data identified several candidate DUX4-interacting proteins. The goal of our proposed studies is to identify critical interactions between DUX4 and its candidate binding partners that we can therapeutically target to abolish the toxic effects of DUX4 in FSHD muscles. Our proposed aims will define DUX4 protein binding partners and mechanisms, and delineate the influence of protein-protein interactions on the DUX4-associated pathogenic cascade. We plan to pursue the following two specific aims to test our hypothesis: Specific Aim 1: To define the binding partners and protein-protein interaction mechanisms of the DUX4 C-terminal domain. Specific Aim 2: To examine the functional significance of protein-protein interactions of DUX4 that are critical for DUX4 toxicity.
Summary (Provided by Applicant): The recent discovery of DNA-binding factors whose sequence specificity is encoded by modular domains that recognize single bases (TALENs) or by a guide RNA (CRISPRs) have opened up tremendous new possibilities in genome editing. With early support from a 2 year ARRA grant, and now with continuing support of an NIH R01, we have developed a zinc finger nuclease (ZFN) that targets 4q35.2. We have used this tool to introduce a new telomere at this site in FSHD iPS cells, which effectively eliminates the genetic lesion. Individuals who lack 4qter on one allele are normal, and our targeted iPS cells that have lost the contracted D4Z4 element are similarly normal. This modification rids the cells of DUX4 mRNA expression and corrects a differentiation defect that we have identified in FSHD iPS cells. We seek funding from the FSH Society to expand this research program (1) to include FSHD human embryonic stem cells (our NIH grant supports FSHD iPS cells), (2) into the exciting new area of CRISPR technology with more specific genetic reversion of the pathogenic 4qA161 allele, and (3) to test the hypothesis that epigenetic silencing can be introduced by targeting D4Z4 with engineered sequence-specific chromatin nucleators. Aim 1. To correct the FSHD locus in human embryonic stem cells bearing the FSHD mutation. Our work to date has shown that FSHD iPS cells express DUX4 mRNA and suffer from an impaired response to Pax7-induced skeletal muscle differentiation and that these phenotypes are reverted by genetic removal of the contracted D4Z4 array. These iPS cells were derived from myoblasts, therefore there is some question of whether these phenotypes represent an epigenetic memory of the pre-iPS cell type. It will therefore be essential to perform this genetic correction in FSHD human embryonic stem cells. Aim 2. To design CRISPRs that target existing and novel sites at 4q35.2. The efficiency of targeted integration with our ZNF reagent is low, therefore we will test whether our existing genetic repair method can be made more efficient by CRISPER technology. We will also design and test CRISPERs targeting the pathogenic poly A signal, which may allow correction of the locus without elimination of the entire D4Z4 array. Aim 3. To use engineered sequence-specific DNA-binding tools to target a chromatin nucleation complex to D4Z4. While most enthusiasm about TALENs and CRISPRs has been around their ability to target a nuclease to introduce double strand breaks in DNA, they can also be used to target other proteins to DNA. Because FSHD is caused by inappropriate relaxation of D4Z4 chromatin on the contracted allele, we will attempt to reestablish heterochromatin at this site by fusing an engineered D4Z4- specific DNA-binding domain to proteins involved in nucleating heterochromatin. These studies take advantage of and leverage an existing research program in genome editing of FSHD iPS cells, and will provide the field with valuable new tools to study the pathogenesis of FSHD, and to develop cell therapies based on corrected, isogenic, iPS cells. Dr. Kyba’s project is jointly funded through the Society as a result of collaboration and partnership with The FSHD Canada Foundation, Calgary, Alberta.
Summary (Provided by Applicant): In the last years, most of the efforts in the research on Facioscapulohumeral Muscular Dystrophy (FSHD), the third most common form of muscular dystrophy, have been focused on the characterization of the non-conventional genetic mechanism activated by pathogenic D4Z4 repeat contractions that underlies the disease. In our study, we will make use of microdialysis with high cut-off membranes, a technique that has never been applied to the study of skeletal muscle, with the aim of elucidating the pathogenetic mechanisms downstream the genetic lesion, with a particular focus on the poorly clarified inflammatory aspects. In the view of a translational research where information coming from clinical imaging is merged with molecular data, we will perform a comparison between the microenvironment of affected muscles in early disease stages (STIR hyperintense and T1-weighted normal signal on muscle MRI, i.e. muscles showing oedema changes but not yet or only partially replaced by fat tissue) and the microenvironment of apparently unaffected muscles (normal T1-weighted and STIR signal on muscle MRI), in FSHD patients. Muscle microdialysates will be analysed using xMAP technology (multi-analyte profiling beads) to compare the levels of inflammatory cytokines, chemokines and growth factors in the two conditions and to characterize the pattern of inflammation and mediators involved. This will allow a better understanding of the role of the inflammatory process in the disease, the identification of biomarkers of disease activity at single muscle level and, finally, the acquisition of information useful for the development of a targeted anti-inflammatory therapy. In the future, the high cut-off muscle microdialysis protocol could be used for molecular monitoring and eventually drug administration in neuromuscular disorders.
Summary (Provided by Applicant): Facioscapulohumeral muscular dystrophy (FSHD) is the third most common myopathy worldwide, but its prevalence may have been underestimated, with it being the commonest muscular dystrophy in Europe. FSHD is an adult-onset, autosomal dominant disorder characterised by wasting of facial muscles and upper body musculature. Disease can progress to affect muscles of the lower extremities and can severely impair quality of life. Over 95% of FSHD cases are caused by contraction of the D4Z4 microsatellite repeat on the subtelomeric region of chromosome 4. In unaffected individuals the D4Z4 repeat region comprises 11-100 D4Z4 units, but in FSHD patients the number is reduced to less than 11. At least one D4Z4 unit is required to cause FSHD however, and only when inherited with a specific polymorphism on the distal end of chromosome 4 (4qA161). Each D4Z4 unit contains an open reading frame for the double homeobox 4 (DUX4) gene, with the 4qA161 polymorphism providing a polyadenylation signal for DUX4 transcripts generated by the last D4Z4 unit. This permissive chromosomal configuration generates stable DUX4 transcripts and it is likely that FSHD is caused by a toxic gain-of-function of elevated levels of DUX4. Little is known about the function of DUX4 and the challenge is now to elucidate how elevated levels of DUX4 cause muscular dystrophy. DUX4 is a putative transcription factor; its N-terminus contains two homeodomains with high similarity to the homeodomains of the transcription factor PAX7, and the C-terminus is an activator of transcription. DUX4 induces apoptosis, with lower expression levels decreasing MyoD and affecting myoblast function and differentiation: important as myoblasts and myotube formation are compromised in FSHD. Many gene expression studies have been performed on FSHD, and other muscular dystrophies, but these have not been thoroughly analysed by the latest bioinformatic techniques. To address this, we developed a novel differential network methodology, designed to identify perturbed signaling pathways in disease networks from just such expression data. Using this novel mathematical methodology, we performed a meta-analysis of multiple publicly available gene expression data sets from FSHD muscle biopsies. We then removed changes associated with muscle wasting, aging, atrophy and inflammation following meta-analysis of appropriate data sets. This integrated output is a high-confidence unified network of pathway changes explaining FSHD pathomechanisms. Interrogation of our network has revealed many promising drug targets and candidate therapeutics. Here, we wish to extend and refine this analysis to identify pathways in our FSHD network that lead to compromised muscle repair and regeneration. To address this, we will collect and analyse by RNA-seq, a high-frequency time course of genome wide gene expression during myogenic differentiation in FSHD. Using mathematical methodologies with optimised network theoretic tools on this gene expression dataset, will reveal molecular mechanisms of myogenesis in FSHD. We will also perform high-frequency time-course RNA-seq on myogenic cells ectopically expressing DUX4, with the aim of identifying primary DUX4 transcriptional-mediated gene expression changes. This will order our FSHD network to identify causal signaling events in FSHD, further eliminating non-specific general adaptations to muscle wasting, inflammation, disuse etc. It will also better identify pathways in the network directly linked to DUX4. Together these results will identify key pathway targets, modification of which should help restore muscle regeneration in FSHD, reversing muscle weakness and wasting. Our ultimate aim is to gain knowledge on FSHD myogenesis and inform the design of therapies for FSHD. There are four objectives: 1. Obtain the first high-frequency time course of genome-wide gene expression during myogenic differentiation in FSHD and control human myoblasts. 2. Obtain a high-frequency time course of genome-wide gene expression in myoblasts expressing ectopic DUX4. 3. Apply our mathematical methodologies with optimised network theoretic tools for analysis of time course gene expression datasets generated. 4. Validate a selection of identified targets for rapid translational studies as potential therapeutics for FSHD.
The FSH Society has allowed us to expand our focus to include a novel technique to measure muscle structure. Electrical impedance myography (EIM) is a fast, non-invasive way to obtain quantitative information about muscle structure which may correlate with motor strength and function in FSHD. The device, manufactured by Convergence Medical Devices (Figure 1, Boston, MA), uses a low-intensity electrical current to obtain information about underlying muscle structure by taking advantage of the relationship of muscle structure to the impedance of current flow through the muscle. EIM is well suited toward investigation of muscles important to FSHD but not easily testable by traditional strength measures, including facial, abdominal, and paraspinal muscles. The funding from the FSH Society makes projects like this possible. The hardest part of any project is getting money to get the projects off the ground: the FSH Society grant has enabled us to start recruiting participants to test these outcomes, and allowed us to apply for additional funding from organizations like the National Institutes of Health to complete this project. It is of vital importance for the FSHD research community that development of outcome measures parallels advancements in molecular pathophysiology and drug development. At the completion of this project we expect to have three valuable new FSHD-related outcome measures for future clinical trials.
The goal of the project is to develop an accurate and robust diagnosis for FSHD. Although FSHD is reported to have a one in 20,000 incidence, there is great concern that the actual number of affected individuals is significantly higher due to undiagnosed cases (with a likely incidence of 1/7,000). Most cases of FSHD involve mono-allelic deletion of macrosatellite D4Z4 repeat sequences at the subtelomeric region of chromosome 4q (4qter D4Z4) (FSHD1), while the remaining <5% of cases demonstrate no D4Z4 repeat contraction (FSHD2). Proper diagnosis depends initially on recognizing clinical signs and symptoms, and differentiating FSHD cases from other muscular dystrophies. Molecular studies have been used to reinforce the clinical impression. The primary approach is detection of 4qD4Z4 repeat contraction by pulsed-field gel electrophoresis (PFGE). However, this method cannot identify phenotypic FSHD2, and certain band patterns can prove difficult to interpret. Therefore, we have urgent need for a better diagnostic technology. The Yokomori group previously found a specific change in histone modification (histone H3 lysine 9 trimethylation (H3K9me3)) at the D4Z4 repeat sequences that is detected in both FSHD1 and FSHD2 patient cells. Importantly, this change is highly specific for FSHD, and is seen also in patient cells from blood samples. Thus, in this project, we plan to test the possibility of using detection of the loss of H3K9me3 in patient chromatin as a diagnostic method for FSHD. We plan to use peripheral blood mononucleocytes (PBMCs) from patient blood samples. Detection of H3K9me3 loss will be assessed by a biochemical method (chromatin immunoprecipitation (ChIP)). We will assess the specificity of our protocol by testing blood samples from healthy individuals, from patients of different ages and disease severities, and from individuals with unrelated muscular dystrophies or unrelated diseases. Related previous grant: Epigenetic abnormality in FSHD Investigators. Weihua Zeng, Ph.D. and Kyoko Yokomori, Ph.D., University of California, Irvine, California. From February 2011: $8,875 for three-month extension.
2012 grant awards
Facioscapulohumeral muscular dystrophy (FSHD), the third most common myopathy, is an autosomal dominant neuromuscular disorder characterized by progressive weakness and atrophy affecting specific muscle groups. FSHD is not due to a mutation within a protein-coding gene, but is caused by contraction of the 3.3 kb macrosatellite repeat D4Z4 in the subtelomeric region of chromosome 4q35. While there is general agreement that D4Z4 deletion leads to over-expression of 4q35 genes, the molecular mechanism through which D4Z4 regulates chromatin structure and gene expression is poorly understood. Consequently, no therapeutic tool to control the aberrant 4q35 gene expression in FSHD is currently available. Polycomb (PcG) and Trithorax (TrxG) group proteins act antagonistically in the epigenetic regulation of gene expression and they play crucial roles in many biological aspects such as development, cell proliferation and cancer. In Drosophila, PcG and TrxG proteins bind to specific DNA regions termed Polycomb/Trithorax Response Elements (PREs/TREs), constituting a regulated switchable element that influences chromatin architecture and expression of nearby genes. D4Z4 shares several features with PREs/TREs. Indeed, my previous results (Cell 2012 149:819-31). showed that Polycomb group of epigenetic repressors targets D4Z4 in healthy subjects. Furthermore, I found that Polycomb proteins are required to maintain 4q35 genes repressed and that D4Z4 deletion is associated with reduced Polycomb silencing in FSHD patients (Cell 2012 149:819-31). My preliminary results strongly suggest that D4Z4 could be the first PRE involved in a human genetic disease. An attractive hypothesis would be that a D4Z4 copy number above the threshold of 11 repeats is able to stably substain a Polycomb-mediated repression of 4q35 genes, while few copies of the repeat fail to do this efficiently. Here, I propose to rigorously investigate the PRE activity of D4Z4. These studies will allow a deep understanding of the D4Z4 mechanism of action and will lay the basis to develop therapeutic approaches aimed at normalizing aberrant 4q35 gene expression in FSHD. My specific aims are: 1.) To understand the mechanism through which the deletion of D4Z4 repeats below a threshold copy number is affecting 4q35 gene expression in FSHD; 2.) To identify potential therapeutic targets.
The genetic and biological events that result in Facioscapulohumeral muscular dystrophy (FSHD) pathogenesis are complex and the link between the genetic aberration and manifestation of symptoms is still elusive. We hypothesize that there might be cellular and genetic alteration in the early stage of myogenesis in FSHD patients. The establishment of human induced pluripotent stem cells (hiPSCs) ushered a new era in biomedicine and can be useful for modeling pathogenesis of human genetic diseases, autologous cell therapy after gene correction, and personalized drug screening. Our lab has been studied human genetic disorders by using induced pluripotent stem cells (hiPSCs) that is a new type of stem cells without destruction of any embryonic tissues or embryos. In addition, we already built a novel methodology in highly innovative manner to directly derive and prospectively isolate skeletal muscle from the hiPSCs. Here we propose to establish hiPSC lines from FSHD patient’s somatic cells. Our proposed study will enable us to isolate FSHD-specific skeletal muscle cells for better understanding of FSHD pathogenesis in human system as well as potential autologous cellular therapy accompanying with genetic correction in near future.
Our previous study showed that DUX4 was up-regulated in patient’s muscles of FSHD and transcriptionally regulated paired-like homeodomain transcription factor 1 (PITX1). The muscle-specific expression of Pitx1 in transgenic mouse model showed muscular dystrophy phenotype similar to FSHD [Pandey et al., 2012]. Expression profiling data of Pitx1 transgenic mice showed that 16 major autophagy genes, including damage-regulated autophagy modulator (Dram1) were mis-regulated in the muscle over-expressing PITX1. To determine whether the autophagy pathways were also affected in FSHD, we investigated the autophagy state in FSHD myoblasts as well as patients’ muscle biopsies. Our data showed disease-specific up-regulation of a master autophagy regulator, DRAM, in FSHD muscle biopsies but not DMD or controls. To further characterize the autophagy state in FSHD myoblasts we cultured the myoblast in differentiation media and we found that DRAM was up-regulated in FSHD myoblasts compared to the control myoblasts. We then examined two proteins critical to autophagy activities, p62 and LC3B. The p62 protein binds both ubiquitinated substrates and LC3B [Pankiv et al., 2007], and has been used as an indicator of autophagic flux. In addition, the accumulating of p62 has been used as an indicator of defective autophagy [Settembre et al., 2008; Ju et al., 2010]. In our study, instead of down regulation when autophagy is activated, p62 showed up-regulation in FSHD myoblasts suggesting a defect in autophagy activation. We further checked the LC3B-II to LCB3-I ratio (LC3B-II/I) which is a commonly used marker for autophagy activation. Because LC3B-II is formed only when autophagosomes are generated, the LC3B-II/LC3B-I ratio represents the density of autophagosomes in cells. The significantly lower LC3B-II/LC3B-I ratio in the FSHD myoblasts indicated again a suppression of autophagy in the myoblast. The suppression of autophagy is also supported by accumulation of ubiquitinated protein in the FSHD cells. While the activation of DRAM should activate the downstream autophagy pathways, we observed a defect in autophagosome formation. Interestingly, the up-regulation of LAMP1 and 2 at mRNA level in muscle biopsy of patients with FSHD suggests that the lysosomal system is activated and ready for the later steps of forming autophagolysosomes. However, the autophagy process is somehow disrupted in FSHD myoblast.In addition, the aberrant expression of DUX4 is a cause of FSHD so we would like determine whether defect in autophagy process is directly linked with expression of DUX4. On the basis of our preliminary result we hypothesize that defect in autophagy causes differentiation defect in myotubes formation in FSHD. In addition, autophagy defect is directly induced by aberrant expression of DUX4. In proposed study, we will examine the expression changes of the key regulators of autophagy and further investigate the mechanisms involved in autophagy defects in FSHD. In addition, we will knock-down the DUX4 expression in the FSHD myoblasts to determine whether the autophagy defects are directly induced by the aberrant expression of DUX4 in the cells.The goal of this current proposal is to understand the mechanism and to identify molecular pathways for treatment development. The aim for this study as follows: Aim 1: To determine the expression of DRAM, p62, Autophagy related gene 5 (ATG5), Autophagy related gene 4B (ATG4B), LC3B, and LAMP1 in patients with FSHD. We anticipate that DRAM, p62 and LAMP1 will show higher expression whereas LC3B-II/LC3B-I ratio will be low in FSHD. Aim 1A: To determine the expression of DRAM, p62, ATG5, ATG4B, LC3B, and LAMP1 in FSHD myoblasts with and without autophagy induction. Aim 1B: To determine the expression of DRAM, p62, ATG5, ATG4B, LC3B, and LAMP1 in muscle biopsies of patients with FSHD. Aim 2: To determine whether the defects in autophagy are due to inhibition of fusion between the autophagosomes and lysosomes. We anticipate a reduction in fusion of lysosome to autophagosomes will be observed in FSHD myoblasts but not in the control myoblasts. Aim 2A: To determine the inhibition of fusion efficiency between the autophagosomes and lysosomes in immortalized FSHD myoblasts. Aim 2B: To determine the expression of ATG4B, a key regulator of LC3B conversion, in immortalized FSHD myoblasts. Aim 3: To determine whether the defects are induced by DUX4 by knocking down DUX4 in the immortalized human myoblasts using antisense oligonucleotides against DUX4. Aim 3A: To determine the expression of DRAM, p62, ATG5, ATG4B, LC3B in proliferating and differentiating myoblasts after knocking-down DUX4. Aim 3B: To determine whether DUX4 knock-down can correct the inhibition of fusion between the autophagosomes and lysosomes in FSHD myoblasts.
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common forms of muscular dystrophy with an estimated prevalence between 1:15,000 and 1:20,000. The clinical spectrum of disease severity is wide, and the regional distribution of muscle weakness, as well as the pattern of progression, is unique. The molecular defect in FSHD on chromosome 4q35 was described in 1992 but the molecular pathophysiology remained unknown until recently. A unifying model has now emerged proposing the aberrant reactivation of the DUX4 gene—resulting in a toxic gain of function- in the pathophysiology of FSHD. This FSHD model has provided, for the first time, therapeutic targets for FSHD, and it is expected that several potential therapeutic interventions will emerge in the coming years. Because of these recent discoveries, there is an urgent need to develop the tools necessary to effectively and efficiently conduct therapeutic trials in FSHD. Existing validated outcome measures in FSHD are neither sensitive to change nor intuitively patient-relevant. More sensitive outcome measures are needed for a more efficient drug development process. The need for patient-relevant outcome measures was emphasized in the proceedings of the 2010 FSHD European Neuromuscular Centre meeting. Moreover, there is increasing emphasis by the FDA on the development of outcome measures that are clinically meaningful and based on the patient’s perspective. There are currently two validated, commonly utilized outcome measures in FSHD (manual muscle testing [MMT] and quantitative myometry) both of which are based on direct strength testing. Although direct measurement of muscle strength makes intuitive sense in a myopathy, what minimum change in such a measure can be considered clinically relevant is not clear. There are, additionally, two FSHD-specific clinical severity scales that have been validated in cross-sectional studies; however, neither the responsiveness to change over time nor the direct relevance to patients has been demonstrated. Moreover, as 10 and 15 point ordinal scales, they are not likely to be highly sensitive to change. Here we plan to test the reliability, validity and responsiveness to change of two FSHD-specific outcomes: the FSHD Health Inventory (FSHDHI) and the FSHD Functional Outcome (FSHD-FO). Both of these instruments were developed based on direct patient input to reflect the most prevalent and important physical limitations of FSHD. We will recruit 35 participants with FSHD for 4 visits over one year of follow up. Outcomes will be compared at baseline and longitudinally to traditional measurements such as the composite MMT score, existing FSHD clinical rating scales, and SF-36 health survey. Additionally an anchoring technique will be used to determine the minimally clinically important change. We expect that this proposal will provide preliminary data on the utility, ease of administration, reliability and validity, and responsiveness to change over one year of two novel and clinically relevant FSHD-specific outcome measures. We have designed our budget so that reliability and convergent validity are tested in year 1; and responsiveness in year 2, months 12-18. It is of vital importance for the FSHD research community that development of outcome measures parallels advancements in molecular pathophysiology and drug development. The scales presented here both represent valuable, patient-relevant tools for the FSHD clinical trial toolkit.
This proposal outlines a postdoctoral fellowship project, which I (AK Zimmermann) am planning to conduct in the laboratory of Francoise Helmbacher at IBDML in Marseille, France over the next two years. I arrived in the lab 5 months ago and have already initiated the research project described below (see preliminary data). The Helmbacher laboratory studies mechanisms that contribute to assembly of neuromuscular circuits during development and associated pathologies. Recently the lab has begun work on facioscapulohumeral muscular dystrophy (FSHD), a devastating human disease characterized by degeneration of muscles in the face and shoulder area. Mechanisms contributing to FSHD remain elusive although pathogenic deletions within a D4Z4 macrosatellite array on chromosome 4 have been identified in the majority of clinical cases. Several studies investigating how this genetic alteration exerts its pathogenic effect have led to propose a contribution of deregulated expression of genes proximal to the deleted region (FRG1 & 2, ANT1), as well as a toxic effect of DUX4, a transcription factor whose transcription is enabled in the pathogenic context. However, a role for these genes in the selective muscle deficits associated with FSHD has not been irrevocably established and therefore additional candidates remain to be identified. The Helmbacher laboratory has shown that FAT1, a protocadherin gene also located near the D4Z4 locus, regulates muscle development and may play a role in physiology of mature skeletal muscles as well. In two mouse mutant models, FAT1-deficient mice were found to develop an FSHD-like phenotype, including both muscle and non-muscle defects. Notably, the developmental abnormalities of muscle shape appear to prefigure the map of muscles affected in FSHD patients. In addition, analyses of human foetal biopsies suggested that tissue-specific silencing of FAT1 might be a causal mechanism in FSHD. This was supported by the finding that several FSHD patients without the classical D4Z4 abnormality carried a deletion of a cis-regulatory enhancer of the FAT1 gene. Here, I propose to study the role of FAT1 in FSHD pathogenesis, and specifically to answer the following questions: 1. Owing to a conditional allele of FAT1 developed in the Helmbacher lab, I will use a tissue-specific ablation approach to ask in which tissues FAT1 expression is relevant for proper muscle migration (candidates include muscle, nerve, vascular and connective tissues) 2. Using primary culture assays on myoblasts and myotubes isolated from Fat1 mutant embryos, I will ask whether FAT1 also play a role in muscle function, as suggested by its subcellular association to the t-tubule excitation-contraction coupling system. Ultimately, alterations of these functions in FSHD patients might be accessible to preventive therapies. 3. Finally, I will ask how FAT1 silencing contributes to dysregulation of retina vasculature, a symptom of FSHD that may provide clues about the molecular mechanisms associated with both muscle and non-muscle phenotypes of disease? Ultimately we hope these strategies will contribute to the development of therapeutic targets aimed at bypassing FAT1 silencing in FSHD and maintaining functional Fat1 levels in muscle prior to worsening of the muscle degeneration symptom.
The most critical need in the FSHD field is a reliable and faithful mouse model of FSHD. This has been inhibited in the past by lack of a consistent and consensus understanding of the gene misregulation in the human condition that leads to FSHD pathology. Now that there is wide spread agreement about the involvement of DUX4-fl in FSHD pathology there are different barriers; the severe cytotoxicity of DUX4 and its lack of conservation in mammals. As such, the field has so far failed to generate a genetic mouse model based on DUX4 expression that recapitulates the DUX4-fl expression profile and FSHD-like pathophysiology. This project proposes to generate a regulable and tunable strain of D4Z4/DUX4 transgenic mice using the Cre/lox system and targeted transgenesis into the Rosa26 locus. Importantly, this model incorporates the downstream cis regulatory elements and DUX4 splicing and polyadenylation of the FSHD-associated 4q35 locus. This is different from any of the mouse models discussed at meetings (none are published) that fail to show any phenotype. The targeting construct has already been generated and shown to function properly in human and mouse myogenic cell culture and myotubes. With this construct we believe we can manipulate DUX4-expression in mice 1) to a range of cells in a population (1:50 down to 1:5000) in the developmental profile of DUX4 expression and/or 2) in any select tissue or spacio-temporal pattern desired. These mice will prove invaluable for therapeutic screening and understanding DUX4 function. As such, once generated and initially characterized we will make these mice available to the FSH community at large in a timely manner for those with therapeutic approaches.
FSHD is characterized by an asymmetric progressive weakness and wasting of the facial, shoulder and upper arm muscles. Hearing loss and retinal vasculopathy are frequently accompanied. Accumulating evidence supports the hypothesis that derepression of DUX4, a double homeobox gene located within D4Z4 unit in chromosome 4q35, play a role in the pathogenesis of FSHD. However, a major problem with this hypothesis is the extremely low abundance of DUX4 expression in FSHD muscle. It has been shown that approximately one cell per 1000 expresses DUX4 protein in cultured FSHD muscle cells. How this sporadic burst of DUX4 expression can cause a chronic and progressive myopathy is largely unknown. To address this question, generation of DUX4 animal model is essential. However, there is currently no good animal model due to the toxic nature of DUX4 where overexpression of DUX4 induces apoptosis to many types of cells, resulting in embryonic lethality. This has hampered the further understanding of FSHD pathogenesis and the development of therapeutic approaches. To generate DUX4 animal model, we injected extremely small amount of human DUX4 mRNA into zebrafish embryos. Microinjection of 0.2 or 0.1 pg of DUX4 mRNA (≈ 1 x 105 copies) caused asymmetric abnormalities on their eyes and fins. Injected embryos also showed affected muscle birefringence and slow swimming, suggesting muscle degeneration and weakness. These phenotypes are very similar to those observed in FSHD patients. We believe that DUX4 injected in small amounts into zebrafish is a good animal model for investigating the pathogenesis of FSHD and the impact of DUX4 on development. In this proposal, we plan to define the phenotype of the DUX4 mRNA injected zebrafish model to determine how much and where in development expression of DUX4 can cause the human FSHD-like phenotype in zebrafish. Furthermore, we plan to develop a conditional DUX4-transgenic zebrafish to create stable model. These zebrafish models will help us to understand DUX4-mediated pathogenesis in vivo, and provide us a platform to screen a number of small molecules for therapeutic approach.
FSHD is an autosomal dominant pathology recently ranked to the most prevalent muscular dystrophy. The genetic locus of the FSHD pathology has been identified 20 years ago, but the molecular mechanisms leading from this genotype to FSHD are still not clearly understood. Indeed, despite recent findings which highlighted the notion of permissive chromosome for FSHD, and the fact that Dux4 is always expressed in FSHD biopsies from these permissive chromosomes, the consequence of this expression on muscle development and function is not well established and the link between Dux4 expression and the development of FSHD pathology is not clearly understood. This reinforces the complexity of FSHD and emphasizes the need to identify other genetic elements putatively involved. Recently, in a collaborative effort involving 3 French laboratories (Francoise Helmbacher, Nicolas Levy and ours), we have identified a new gene named FAT1 which, when down-regulated in mice, recapitulates FSHD muscular phenotypes: at early post natal stages, shoulder and face muscles present an asymmetric atrophy, whereas at later stages, a widespread muscular dystrophy is observed. Moreover, the FAT1 mutant mice also present some non muscular FSHD characteristics such as a retinal vasculopathy. In human, FAT1 is located in 4q35 and belongs to the Planar Cell Polarity (PCP) family which is involved in coordinating tissue polarity, morphogenetic movements, and polarized cell flow. Interestingly, we have also observed that FAT1 mRNA is systematically down-regulated in human FSHD fetal muscle biopsies (but not in brain) as compared to age matched control fetuses. Moreover, in several FSHD2 human samples which do not present the typical FSHD contraction, deletions in the FAT1 gene have been found. All together, these observations strongly suggest that FAT1 may play a major role in the FSHD pathology. Since FAT1 has never been described to play a role in muscle physiology in mammals, our goal is now to understand its biological function and to analyze how it may underlie the onset and progression of FSHD disease. We have already demonstrated that different FAT1 isoforms co exist in human muscle cells and that they are differentially expressed in proliferating and differenciating conditions. Moreover we have also observed that the expression of some isoforms can be coregulating each other, thus highlighting the complex regulation of FAT1 expression and the urgent need to decipher its specific expression. Our aim is to specifically up regulate or down regulate each isoform in normal or FSHD fetal myoblasts and to analyse the effects on myotube formation, on the expression of the other isoforms, on the localization of the protein using FAT1 specific antibodies and on the modulation of expression of other factors we have recently identified in biopsies and in cell cultures (proteins, miRNA), which represent putative new biomarkers for FSHD. Finally, different miniFAT1 will be cloned in viral vectors which will be used to transduce the FAT1 mutant mice in order to try to rescue the FSHD phenotype.
Facioscapulohumeral muscular dystrophy (FSHD) is genetically caused by the contraction of D4Z4 DNA repeats located on chromosome 4 in 4q35. Although the genetic defect was identified 20 years ago, the exact molecular mechanism causing the disease is unknown. Because of the unique nature of human D4Z4 repeats, there is currently no mouse disease model. To provide such a valuable tool, we have developed a humanized mouse model for FSHD. This model was obtained by the engraftment of FSHD patient derived myoblasts into mouse muscle. Because of the dominant nature of the disorder, we hypothesized that the FSHD engrafted fibers will display a disease phenotype and recapitulate pathological molecular mechanisms associated with FSHD that will allow us to study the development of the disease. Our preliminary work has established the feasibility of this project. Our findings demonstrate the successful, high efficiency engraftment of myogenic cells from FSHD and control subjects into injured, regenerating tibialis anterior (TA) muscle of immune-compromised mice. Early passage myoblast cells from cohorts of FSHD probands and their appropriate controls (i.e., a first degree relative) for these studies were provided by the unique cell repository of the Boston Biomedical Research Institute (BBRI) Wellstone Cooperative Research Center for FSHD Research. We have grafted these standardized cultured cells into mouse muscle to obtain the FSHD humanized mouse model, thereby generating a well-controlled in vivo model for the study of FSHD. We here propose to investigate FSHD disease progression in the humanized mouse model, through studies of the expression of DUX4, an FSHD candidate disease gene, and a larger set of 143 putative FSHD disease biomarkers, which includes a number of DUX4 target genes, identified by the BBRI Wellstone Center by microarray analysis of differentiated FSHD and control myogenic cells. Furthermore, in vivo animal imaging technologies will be utilized to investigate the survival, regenerative capacity and maintenance of FSHD myogenic cells engrafted into injured mouse TA muscle as a pathological mechanism for FSHD In summary, this work will contribute to the understanding of the FSHD pathogenesis in vivo by defining the cellular and molecular disease pathology of FSHD using the unique humanized mouse model of FSHD. Research findings will enable identification of new drug targets for FSHD treatment and provide an animal model for preclinical studies of RNA silencing and small molecule FSHD drugs.
FSHD is an autosomal dominant neuromuscular disorder, with an incidence of 7: 100,000 recently ranked as the most common and prevalent rare muscular dystrophy of the adult. The number of D4Z4 is a critical determinant of the age of onset and clinical severity of the disease. A region distal to D4Z4 generates two allelic forms, 4qA and 4qB both equally common in the general population but FSHD is mainly associated with the 4qA allele. In addition, other polymorphisms, qualified as permissive alleles, exist; the pathology being often associated with the 4qA161 allele. In 5-10 % of families with a typical FSHD phenotype, there is no linkage to 4q35 and this type is referred to as type 2. Over the last decade, major advances have occurred in the understanding of the genetics of this disorder however the exact patho-mechanisms secondary to the genetic defect are still not understood. The DUX4 ORF is localized within D4Z4 and since at least one repeat is necessary to generate a pathogenic phenotype it has been hypothesized that DUX4 overexpression contributes to the pathology by leading to the production of a toxic protein in 1 in 1000 muscle nucleus. While several groups are actively seeking targets of the DUX4 protein, a technically challenging approach, our goal is to understand what leads to DUX4 expression. The deletion of repetitive elements and changes in epigenetic marks across the D4Z4 array such as DNA hypomethylation or decrease in H3K9 trimethylation also indicates that FSHD involves chromatin changes and epigenetic alterations. Interestingly, a localization of the 4q telomere at the nuclear periphery has been reported including by our group (Ottaviani et al., 2009; Arnoult et al., 2010), in close proximity to heterochromatin suggesting that subnuclear positioning contributes to this peculiar pathology. However, the links between epigenetic changes, nature of the 4q35 sequences, DUX4 expression and muscle phenotype have never been fully demonstrated. Therefore, based on our current knowledge and expertise, we are deciphering the link between the subnuclear positioning of the D4Z4 array, epigenetic changes and DUX4 expression in the pathology. Thus, we focus on the regulation of the 4q35 region as a whole through the constitution and exploration of a unique cohort of patients’ samples including atypical cases and cellular models. The strategies proposed here, would lay new grounds for the deciphering of this complex disorder. • Within the frame of this project, a cohort of samples from patients is available including accompanying detailed patient histories and genotyping. • Primary myoblasts and fibroblasts for FSHD pathogenesis, • Induced pluripotent cells (iPSCs) from FSHD1 and 2 patients. This cellular model will be a very valuable tool to investigate the role of epigenetic changes in the tri-dimensional organization of the FSHD locus and the regulation of a putative “FSHD gene” during differentiation and investigate how DNA methylation contributes to the pathogenesis. The epigenetic mechanisms regulating the 4q35 locus remain poorly understood, including how DNA methylation is controlled in the pathology and whether hypomethylation is a cause (increased instability) or a consequence of D4Z4 array shortening. We aim at understanding whether D4Z4 hypomethylation is an early event that precedes shortening of the repeated array or simply the consequence of the loss of a certain number of repeats by comparing methylation profiles in normal and diseased cells, and investigate the intergenerational transmission of DNA methylation in FSHD1 and 2 patients and correlate this methylation level with disease penetrance, D4Z4 array compaction and DUX4 expression. We have already shown that is it possible to visualize methylated regions through the detection of methylcytosines on combed DNA molecules using anti-m5C antibodies. We wish to use MC combined with DNA methylation detection with the probes designed for genomic analysis in order to evaluate the DNA methylation pattern of the D4Z4 array and flanking sequences for each individual allele on the slide and all D4Z4 copies. The sensitivity and stringency of the method requires further development and tests of different experimental conditions are in progress. Hypomethylation occurs in FSHD2 suggesting that DNA methylation metabolism is globally altered in FSHD. The de novo methyltransferase (DNMT3b) contributes to D4Z4 methylation suggesting that epigenetic programming of the repeat occurs at early developmental steps. We investigated the expression of catalytical DNMTases and co-regulatory splicing variants in myoblasts. Demethylation and chromatin changes might also be subsequent to differentiation. IPSCs provide a unique platform to dissect the mechanisms of epigenetic reprogramming during differentiation for typical FSHD cases and FSHD2. Using this type of model, we are investigating the timing of production of DUX4, and testing whether repeat’s transcription initiates epigenetic regulation in different genomic contexts and vice versa. Gene localization in the nucleus is not arbitrary, it’s a dynamic process and we have previously identified D4Z4 as the first human sequence able to control the localization of its abutting telomere by tethering this chromosome end toward the nuclear periphery. We hypothesize that the tri-dimensional organization of the 4q35 locus is altered in patients, being responsible for deregulation of disease’s gene expression. Using a large collection of patients’ biopsies, we have shown that the expression of several genes located in the 4q35 region is modulated in patients and we are now trying to describe how the tri-dimensional organization of the D4Z4 array might affect gene expression by comparing 4q35 chromatin conformation in patients and unaffected individuals in order to understand the interactions between sequences, either distant or in close proximity during proliferation of muscle precursors and muscle differentiation. To study the tridimensional distribution of DNA sequences within the nuclear volume, we have developed a FISH technique coupled with immunofluorescence in 3D. Since FSHD is a progressive pathology, we have chosen to focus on three different cell types, fetal and adult primary myoblasts and fibroblasts from patients or controls, as well as induced pluripotent stem cells (IPSc) derived from primary fibroblasts. Our preliminary results show that hybridization patterns differ in patients and controls cells. Signal hybridization volumes display a significant difference between patients and controls which might reflect the degree of chromatin relaxation in the pathology. Moreover, signals corresponding to 4q35 probes are more colocalized in patient cells, suggesting an interaction between sequences undergoing the same regulatory mechanisms. Hence, we will consolidate our data by using iPSCs as a model of the pathology. These approaches should bring further insights into the underlying mechanisms of FSHD in order to identify target genes and to understand by which mechanism, a decrease in the number of D4Z4 leads to this muscular dystrophy. This project is expected to lead to a better understanding of the regulation of the 4q35 at the genomic and epigenomic levels. We believe that the different aspects proposed here are required to get further insights into the patho-mechanism of FSHD, a key step for the future development of specific therapeutic targets.
2011 grant awards
D4Z4 repeat array chromatin relaxation and transcriptional de-repression of the non-polyadenylated double homeobox 4 (DUX4) gene unifies D4Z4 contraction-dependent FSHD1 and contraction-independent FSHD2. Only from FSHD-permissive genetic backgrounds the DUX4 transcript originating from the most telomeric unit of the array can be stabilized by a polyadenylation (polyA) signal outside the array. Non-permissive chromosomes fail to stabilize DUX4 in the absence of this polyA signal. Somatic DUX4 derepression in FSHD1 and FSHD2 leads to bursts of DUX4 protein in sporadic nuclei of cultured FSHD myotubes. DUX4 is highly expressed in the germ line. It is low expressed in embryonic stem cells and it subsequently gets silenced during differentiation. FSHD iPS cells fail to silence DUX4 during differentiation. The regulatory mechanisms that act upon DUX4 in muscle are largely unknown and currently we do not know how a protein that is expressed in minute amounts causes a chronic and progressive muscle wasting. While others have used conventional over-expression vectors to study the effect of DUX4, we have consistently observed that using constructs in which the genomic organization of DUX4 is retained, i.e. within the context of D4Z4, the locus creates sporadic bursts of DUX4 expression: not only in FSHD1 and FSHD2 cultured muscle cells, but also in muscle cells cultured from our transgenic L42 mice and in C2C12 cells stably transfected with a genomic D4Z4 construct. These bursts already occur at low frequency in proliferating cells and increase in frequency during differentiation. I aim to identify the epigenetic mechanisms that regulate the bursts of DUX4 activity. I will develop reporter constructs in which the DUX4 ORF in D4Z4 is replaced by a reporter gene but in which otherwise the genomic integrity of the distal DUX4 gene is preserved. These reporter constructs will be used in the following set of experiments: 1. Fluorescent reporter constructs will be used in life cell imaging studies to precisely characterize the bursts of expression. Although the highest somatic expression of DUX4 is observed in differentiated myotubes, occasional nuclei expressing DUX4 can also be observed during proliferation. Life cell experiments will establish whether bursts of DUX4 are cell-cycle dependent or whether other factors regulate DUX4 expression. It will also establish whether a single nucleus can repeatedly express DUX4, or whether this is a one-time event. 2. Inserting a fluorescent reporter in the construct allows for the separation of expressing muscle cells by FACS sorting and comparison of the chromatin structure of expressing and non-expressing cells by ChIP with a panel of histone modifications that allows for the recognition of the major chromatin states in mammalian cells. These chromatin studies will be validated in our extensive panel of primary muscle cells of FSHD patients and controls. I expect this study yield a comprehensive epigenetic map of the FSHD locus in DUX4 expressing and non-expressing cells. 3. The reporter construct will also be used in dedicated and in large-scale screens for compounds that activate or repress DUX4. I will use an established RNA-interference (RNAi) screen (collaboration with Dr. Agami, NKI, Amsterdam) to identify chromatin modifiers that affect the D4Z4 chromatin structure. I will validate these studies in primary muscle cells of FSHD patients and controls. I expect this study to provide mechanistic insight in the chromatin structuring of the FSHD locus in patients and controls. Currently we have identified a uniform molecular mechanism for FSHD. I expect this study to contribute to the current gap in our model of how a protein that is expressed in minute amounts causes a progressive muscle disease. A FSH Society Marjorie Bronfman research grant FSHS-MGBF-019 FSHS-82011-01
Facioscapulohumeral muscular dystrophy (FSHD) is the most prevalent hereditary progressive muscle disorder in humans. It is an autosomal dominant disease that causes wasting and weakness in multiple muscle groups (face, shoulders, and upper arms initially, and legs later in the disease) as well as significant disability in affected individuals. Recent advances in understanding the pathophysiology of FSHD have led to the identification of therapeutic targets. However, we lack appropriate biomarkers that reflect the degree of muscle degeneration and regeneration in these patients. Such biomarkers will be necessary for the successful completion of clinical trials. The proposed study will test the hypothesis that proton magnetic resonance spectroscopy (MRS) and quantitative magnetic resonance imaging (MRI) can be used to define unique metabolic profiles in the skeletal muscle of patients with FSHD and controls with normal muscles. These profiles can then be used as biomarkers of disease severity and surrogate outcome measures in therapeutic clinical trials in FSHD. The initial aim of the project will be to establish quantitative MRS patterns in the skeletal muscles of patients with FSHD. We will accomplish this by performing a cross-sectional imaging study of 30 subjects with genetically-confirmed FSHD and measurable biceps weakness. Each subject will undergo MRI/MRS imaging of the upper extremity, and we will correlate the metabolic profiles of the biceps muscles to disease severity (as measured with quantitative muscle testing). Further specific aims will be developed to: 1.) compare magnetic resonance spectroscopy profiles of subjects with FSHD to groups of healthy and diseased controls, 2.) identify correlations between MRS biomarkers and molecular biomarkers collected from muscle biopsy samples, and 3.) observe longitudinal changes in muscle spectroscopy on repeat studies. The combined output of the proposed project will be an imaging protocol that can be used in future clinical trials in FSHD. A FSH Society Irene Lai research grant FSHS-SLMM-002 FSHS-82011-02
This $12,500 per year grant is supplementary support linked to an NIH R21 1R21NS076671-01 (2-year) application directed towards identifying chemical inhibitors of DUX4. We have previously screened 200,000 compounds and identified approximately 600 inhibitors of cell death induced by DUX4, and current work is directed towards identifying the most promising leads within this set of 600. The NIH application was recently selected for funding and the grant was initiated. This support from the FSH Society will be used to supplement the NIH project, principally by providing additional medicinal chemistry support (allowing us to increase the number of compounds that we follow-up by purchase/synthesis. These compounds help us to understand pathways that can indirectly inhibit toxicity associated with DUX4. See 1R21NS076671-01 [ http://projectreporter.nih.gov/project_info_description.cfm?aid=8225734&icde=10980193 ] for more information on the R21.
FSHD is a muscle degeneration disease genetically linked to contractions of the D4Z4 repeat array on the 4q35 subtelomeric region. Our group has identified the double homeobox 4 (DUX4) gene within each unit of the D4Z4 repeat array and shown that the encoded protein was expressed in primary myoblasts and biopsies of patients with FSHD but not in non-affected individuals. We found that the only stable DUX4 messenger RNAs derive from the last unit and extend to the flanking pLAM sequence that provides a polyA addition signal. This signal is required to develop FSHD as independently confirmed by an eight-laboratory consortium which studied genetic polymorphisms in hundreds of patients and thousands of healthy individuals. In aggregate our collaborative studies with four different groups have shown that the DUX4 protein is a transcription factor that targets a large set of genes, some of which encode other transcription factors that in turn target additional genes. Globally, DUX4 activation at the FSHD locus initiates a transcription cascade leading to muscle atrophy, inflammation, decreased differentiation potential and oxidative stress, the key features of the disease. By differential protein, RNA and gene studies we keep identifying additional FSHD biomarkers and define whether they are direct or indirect DUX4 targets. Strikingly, we found that DUX4 expression in human myoblast induces an atrophic myotube phenotype and atrophy markers. The rationale of our on-going project is that inhibition of DUX4 expression should prevent the global gene deregulation process and allow muscle regeneration in patients. We have first developed small inhibitory RNAs (siRNAs) and conditions to suppress DUX4 protein expression either in primary myoblast cultures transfected with a DUX4 expression vector, or in primary FSHD myoblasts. Addition of DUX4 siRNA to FSHD myoblasts allowed recovery of a normal myotube phenotype with a decrease of atrophy markers. We have started a collaboration with Prof. Steve Wilton (ANRI, Australia) because of his expertise in the exon skipping therapeutic approach with antisense oligonucleotides (AOs) in Duchenne muscular dystrophy. Prof. Wilton provided us with 7 AOs directed against various parts of the DUX4 mRNA characterized in our group: the aim was to either block translation or induce mRNA degradation to prevent DUX4 protein expression. We were able to identify conditions for selective DUX4 inhibition by 3 AOs as done for the siRNAs above in human primary myoblast cultures. Moreover DUX4 mRNA inhibition also affects the expression of several FSHD markers such as μ-crystallin, β-catenin and TP53. These results constitute a proof of concept in myoblast cultures that DUX4 inhibition might reverse the FSHD phenotype. In the present project we want to validate these results by other techniques (RNA and protein expression profiling) and to test the effect of these AOs and siRNA in mouse models in vivo. A FSH Society New York Festive Evening of Music and Song fellowship grant
FSHD is genetically caused by the contraction of D4Z4 DNA repeats located on chromosome 4 in 4q35. Although the genetic defect was identified 20 years ago, the exact molecular mechanism causing the disease is unknown, and there is currently no mouse disease model. To provide such a valuable tool, we will develop a humanized mouse model for FSHD, obtained by the engraftment of FSHD patient-derived myoblasts into mouse muscle. Engrafted human cells are able to form muscle fibers in the host mouse muscle, thus allowing pioneering studies in an in vivo context. Because of the dominant nature of FSHD, we hypothesize that the engrafted fibers will display a disease phenotype and recapitulate pathological molecular mechanisms associated with FSHD that will allow us to study the development of the disease. Our preliminary studies have already established the feasibility of this project. Through the cell repository of the Boston Biomedical Research Institute (BBRI) Wellstone Center, we have the unique opportunity to access early passage myoblast cells from cohorts of FSHD probands and their appropriate controls, i.e., a first degree relative. We will graft these standardized cultured cells into mouse muscle to obtain the FSHD humanized mouse model, thereby generating a well-controlled in vivo model for the study of FSHD. The very pressing issue in the field today is the verification of the current DUX4 model. The humanized mice produced will be used to investigate the hypothesis that DUX4 gene expression is a major cause of FSHD pathogenesis. In the obtained model, DUX4 expression will be evaluated during in vivo regeneration, and the consequence of its expression on fiber turnover and satellite cell renewal will be assessed. This work will contribute to the understanding of the role of DUX4 in vivo, thus providing a better understanding of FSHD pathogenesis. The proposed project will be completed following 2 specific aims: Specific Aim 1: Optimization of the FSHD humanized mouse model. We will improve results obtained in preliminary experiments by designing more efficient transplantation strategies. In order to fully interpret the disease model, we will seek to increase the amount of muscle formed from implanted human cells, by devising more efficient transplantation strategies. The cell repository of the BBRI Wellstone Center provides access to freshly isolated FSHD and their appropriate control muscle cells sorted for CD56 expression, which are expected to have particularly high engraftment potential. However, the timing between the toxin injection and the cell injection, as well as depletion of endogenous satellite cells by irradiation of the mouse legs, may affect the ability of implanted cells to regenerate the murine muscle and will be optimized during this aim. Upon establishment of an effective mouse model, we will look for disease characteristics, as described in Specific Aim 2. Specific Aim 2: Characterization of the FSHD humanized mouse model to evaluate the role of DUX4 during in vivo muscle regeneration. The model obtained in Specific Aim 1 will be characterized by establishing differences between the fibers generated from FSHD cells and fibers from their appropriate control cells in injected muscles. Recent breakthroughs in the field suggest that DUX4, a gene identified inside D4Z4 repeats, expresses a toxic protein in the muscles of patients with FSHD, thus causing the disease. DUX4 may have a normal role during development and the FSHD pathology might involve incomplete developmental silencing of DUX4. However, the precise molecular and cellular mechanisms involving DUX4 remain to be uncovered. The BBRI Wellstone Center, currently investigating DUX4 expression in muscle samples from its cohort collection, has been able to detect DUX4 transcripts in FSHD samples, and these cohorts will be selected for the generation of the humanized FSHD mouse model. Initially, the expression of DUX4 at the mRNA and/or protein levels will be assessed in FSHD- and control transplanted muscles. This will be followed with experiments designed to compare the biological characteristics of the resulting muscle fibers. Finally, we will develop a dynamic approach to investigate the current DUX4 model in following the evolution of the engrafted fiber over time using in vivo bioluminescence live imaging. Murine models surpass in vitro limitations due to their ability to reproduce complex in vivo environment thereby providing a deeper understanding of disease mechanisms. Our model for creating humanized FSHD fibers in murine muscle will recapitulate the mechanisms of pathological fiber formation in vivo, allowing us to fully characterize the disease progression and test potential therapeutic agents. A FSH Society New York Festive Evening of Music and Song fellowship grant
FSHD is considered the most frequent hereditary muscle disorder in adults, affecting one individual in 20,000. It is associated with contractions of the D4Z4 repeat array in the 4q35 subtelomeric region. In non-affected individuals, this array comprises 11-100 tandem copies of the 3.3-kb D4Z4 element while in patients, only 1-10 D4Z4 copies are left (Wijmenga et al., 1992). Our group has identified the double homeobox 4 (DUX4) gene within each unit of the D4Z4 repeat array (Gabriels et al., 1999) and several studies have now demonstrated the causative role of DUX4 in FSHD. We have demonstrate that the stable full-length DUX4 messenger RNA (mRNA) is produced from the last D4Z4 unit in FSHD, using a polyadenylation signal in the flanking pLAM region, located telomeric to the distal repeat (Dixit et al., 2007) as recently confirmed by a study of genetic polymorphisms in hundreds of patients and thousands of non-affected individuals (Lemmers et al., 2010). This polyadenylation site is necessary to develop FSHD on a contracted allele therefore called “permissive chromosome” (Lemmers et al., 2010). The mRNA from this distal D4Z4 unit contain the entire DUX4 open reading frame (ORF) and 1 or 2 alternatively spliced introns in the 3’UTR (DUX4-fl). In addition, a short DUX4 mRNA terminates at the previously described polyadenylation site in the pLAM region but uses a cryptic splice donor site within the DUX4 ORF (DUX4-s). DUX4-fl was only detected in FSHD muscle cells and biopsies, whereas DUX4-s is detected both in control and some FSHD samples (Snider et al., 2010). A long DUX4 mRNA was detected in induced pluripotent stem cells (iPS cells) and human testis where the gene contains 4 additional exons and a more distal polyadenylation signal. Expression of this DUX4 mRNA was suppressed during differentiation of control iPS cells to embryoid bodies whereas expression of full length DUX4 mRNA persisted in differentiated FSHD iPS cells (Snider et al., 2010). These data, together with the conservation of the DUX4 ORF through evolution (Clapp et al., 2007) suggests a possible role of DUX4 in human development. Dr. Tassin intends to undertake a post-doc for three months in 2011 at King’s College London, to initiate a collaborative research project between our lab and that of Dr. P. Zammit. In agreement with Dr. Zammit, our collaborative project will consist of testing antisense oligonucleotides (AOs) directed against the 3’UTR of the DUX4 gene that we have developed in our laboratory, in collaboration with Prof. S. Wilton (ANRI, University of Western Australia). These AOs have undergone preliminary screening in cell culture, but require more extensive testing. Dr. Zammit has developed mouse myofibre models that provide an ideal system to further test our AOs. The satellite cells associated with the isolated myofibres will be infected with retroviral vectors encoding DUX4, and the effects on myogenic progression and apoptosis of AO administration analysed. We want specially to focus on the pLAM region responsible for the stabilisation of the DUX4 mRNA leading to FSHD. This system will allow better understanding of the action AOs, for evaluating their potential suitability as a human therapy. We believe that this collaboration will give us new insights into a potential therapy for FSHD. A FSH Society California Walk and Roll fellowship grant
There is a great need for a valid mouse model for FSHD. Such an animal model would provide a valuable tool for exploring the effects of newly cloned genes and novel proteins on the pathophysiology of this disease. It would also greatly facilitate research towards the development and testing of new therapeutic approaches to FSHD. We propose to examine two possible mouse models of FSHD, the FRG1 over-expressor, from Drs. Davide Gabellini and Rossella Tupler, and mu-crystallin over-expressor, developed by Drs. Patrick Reed and Robert Bloch. I will breed these mice and test them for their physiological and morphological characteristics, and their susceptibility to injury and ability to recover from injury. I will also initiate xenografting studies to create mice with humanized normal and FSHD ankle dorsiflexor muscles, combining methods that are routine in the Bloch laboratory with unique reagents provided by collaborators in the Wellstone Muscular Dystrophy Cooperative Research Center (MDCRC), “Biomarkers for Therapy of FSHD.” These experiments should reveal the usefulness of available transgenic models for the study of FSHD, and promote the development of humanized mouse muscles for the study of the pathophysiology of FSHD in situ. A FSH Society New York Festive Evening of Music and Song fellowship grant
Our preliminary findings indicate that D4Z4 repeat regions indeed interact with other genome regions, and that these interactions are indeed disrupted in FSHD. With a three-month extension of my fellowship, I plan to perform a high-throughput identification of potential target genes that interact with D4Z4 using the recently developed “Chromatin Interaction Analysis using a Paired-End Tag” (ChIA-PET) technique. This strategy enables the genome-wide detection of chromatin interactions mediated by specific factors that are normally assembled at D4Z4. Identification of additional FSHD pathogenic genes other than FRG1 and DUX4 is important to explore future therapeutic targets to improve or prevent the clinical symptoms of FSHD. Previously, with the support from the FSH Society in 2010, we found that a set of factors that normally assemble at D4Z4 repeats do not bind to these repeats in FSHD cells. Interestingly, these factors are known to function in gene silencing and long-distance genomic interactions, which appear to be particularly important for coordinated developmental gene regulation in human cells. Two candidate genes, FRG1 in a neighboring region and DUX4 encoded within D4Z4, have been identified whose artificial over expression did cause muscular dystrophy in vivo or a myoblast differentiation defect in vitro, respectively. The loss of chromatin structure associated with gene silencing at D4Z4 may explain the abnormal expression of these genes in the disorder. However, FSHD patient muscle cells do not always over express these genes. Thus, there are likely to be additional unidentified genes and signaling pathways involved in the pathogenesis of FSHD. Our hypothesis is that D4Z4 normally spreads a silencing effect to target genes through genomic interactions mediated by D4Z4-bound factors. This function is lost in FSHD, resulting in the abnormal over expression of a set of target genes that leads to clinical manifestations of the disorder. I am taking two strategies to test this model; (1) screen for any genes that might have lost factors similar to those that are lost from D4Z4 in FSHD by high-throughput genome-wide chromatin immunoprecipitation (ChIP)-sequencing, and (2) directly search for genomic regions that interact with D4Z4 using biochemical chromatin conformation capture (3C)-related methods. Any candidate genes identified by these assays will be tested for their effect on cell viability, proliferation/differentiation, and muscle-related downstream gene expression. I will try to re-create the expression change detected in FSHD cells in normal human myoblasts (by over expression or repression) and compare it to the phenotypes of FSHD myoblasts to determine whether the candidate gene contributes to the FSHD cellular phenotype. My research aims to decipher the epigenetic abnormality mechanism in FSHD, which should provide novel insight into the disease mechanism and thus potentially present new therapeutic strategies. A FSH Society Sanford Batkin & Helen Younger and David Younger research fellowship grant
We request support from the FSH Society for our pilot project investigating DUX4 expression in unaffected and FSHD subjects. The DUX4-fl expression model for FSHD has not been independently validated, likely due to the lack of quality clinical resources in the field. At this point in FSHD research, validating and expanding upon the DUX4-cytotxicity model for pathogenesis is vital to the entire field and we are best positioned to do the necessary experiments with the unique set of highly controlled reagents being generated by the NIH Wellstone Muscular Dystrophy CRC for FSHD at BBRI. Each Wellstone cohort consists of an FSHD affected subject and an unaffected first-degree relative. Each subject donated two biopsies, one from the biceps and one from the deltoid. A portion of each biopsy was used to derive myogenic cell cultures. Quite surprisingly, in our initial preliminary results using 4 cohorts we found some inconsistencies with the published DUX4 expression results that have warranted further investigation. Therefore we have begun a much larger effort to analyze DUX4-fl mRNA and protein expression in a larger set of Wellstone cohorts using RT-PCR and immunostaining (ICC). However, this project is not funded at all in my lab or in the original Wellstone budget and my lab receives no financial support from the Wellstone Center. The Wellstone has supported us by providing us with cells, which we culture, and RNA which the Louis Kunkel lab purified from biopsies (we do not actually work with the biopsies) and we have been fortunate to receive these Wellstone samples. At this point, to ensure that our results are statistically meaningful, we need to analyze many more cells and biopsy RNAs and it has become cost prohibitive. Therefore I am requesting financial support for consumables and services (DNA sequencing) to conduct these experiments. A FSH Society Cape Cod Walk and Roll fellowship grant
2010 grant awards
We and others have shown that DUX4 is toxic to different cell types, and induces FSHD-associated morphological and transcriptional changes in vitro.As a first step towards developing a targeted therapy for FSHD, we have taken advantage of conditional toxicity of DUX4-inducible myoblasts and we developed a small molecule screening platform for identifying inhibitors of DUX4. In our iC2C12-DUX4 inducible myoblasts, we incorporated full length of the last D4Z4 repeat, so prior its induction, we can not exclude that besides DUX4, some other products are not expressed (RNA, spliced proteins). Assay based on rapid cell death within 24 hours induced by high levels of DUX4 was used for high throughput screen of 200,000 chemicals, part of UT Southwestern HTS compound library. We identified more then 586 compounds with significant rescue ability (60 to >100% cell survival). To identify direct inhibitors, we have conducted serial follow up assays, including secondary screens to eliminate compounds which interfere with the rtTA/TRE inducible gene expression system, to distinguish anti oxidants, to confirm reversion of toxicity in other DUX4-expressing cell types. Several classes of compounds reverted toxicity indirectly, including antioxidants. After these secondary screens, we have narrowed down the list to 82 potentially direct DUX4 inhibitors. The goal of this proposal is to discover a chemical compound/s which efficiently inactivates the DUX4 protein and build on that discovery to develop a drug for a therapeutic approach to FSHD. To achieve this we will have to filter our current list (82 compounds) with additional secondary screens. Among them will be an analysis of MyoD expression and stability as well as cellular localization of the DUX4 protein (Aim 1). We reported that DUX4 is a potent inhibitor of MyoD expression. Therefore, a compound that will rescue MyoD expression in DUX4 induced cells is likely to be a therapeutically effective DUX4 inactivator. We assume that compounds which will be able to inactivate DUX4 in our iC2C12-DUX4 system most likely will be able to rescue FSHD myoblast phenotype. FSHD myoblasts were reported to have impaired differentiation, missregulation of myogenic transcription factors and increased susceptibility to oxidative stress. For that reason, as a functional in vitro study, we will test selected compounds for reversion of FSHD myoblast phenotype (Aim 2). Furthermore, we will test whether selected compounds exhibit their effect on inactivation of DUX4 protein or inhibition of DUX4 transcription or translation (Aim 2). At the end the most potent compound /s will be test for pharmacokinetic and pharmacodynamic properties (Aim 3). The aims of our proposed study target the most crucial topic and urgent needs of FSHD patients: specific and direct pharmacological therapy. Aim 1. To narrow our focus to the most promising direct DUX4 inhibitors. Aim 2. To evaluate effectiveness of DUX4 inhibition Aim 3. To analyze pharmacokinetic and pharmacodynamic properties of the selected compounds.
FSHD was formally classified in 1954, and the primary genetic defect, D4Z4 contraction, was identified in 1992, but the pathogenic mechanisms underlying the disease have only recently started to come into focus. One reason for the difficulties in understanding FSHD biology is the lack of a relevant animal model expressing FSHD-permissive D4Z4 arrays. Since animal models, particularly mice, are crucial tools for studying disease pathogenesis and developing potential therapeutics, the absence of an FSHD mouse model is a fundamental problem in the FSHD field. A major goal of the Harper lab is to generate an FSHD mouse model expressing a single FSHD-permissive human D4Z4 repeat, and to use this model to understand the role of the D4Z4-resident gene, DUX4, in FSHD pathogenesis, and develop RNAi therapeutics targeting DUX4. In preliminary data, supported by previous FSH Society Fellowships to the Harper Lab, we delivered DUX4 to mouse muscle using adenoassociated viral vectors (AAV). DUX4 over-expression in muscle caused myopathy, but DUX4 is generally toxic to many non-muscle cells as well. Thus, we hypothesized that if DUX4 over-expression is an underlying pathogenic event in FSHD, it must be preferentially expressed only in affected muscles. We therefore developed transgenic mice expressing the green fluorescent protein (GFP) gene from the human DUX4 promoter (DUX4p-GFP mice), to determine the tissue and cell specificity of DUX4. In preliminary studies, we observed gross GFP expression in the face, shoulder girdle, and limbs of three independent DUX4p-GFP mouse lines. In this proposal, we will more carefully define the developmental and cellspecific expression patterns of DUX4p-GFP mice, and develop an AAV vector to determine whether a viral-mediated vascular delivery approach can produce the same expression patterns. Ultimately, these studies will be important first steps toward developing an AAV-mediated D4Z4 mouse model. Specific Aim 1: To define the developmental and cell-specific expression patterns of the human DUX4 promoter in mice. Mounting evidence supports the hypothesis that over-expression of the D4Z4-resident DUX4 gene is an underlying pathogenic event in FSHD. DUX4 is generally toxic to many cell types, and since FSHD is characterized by dystrophy of very specific muscle groups, we hypothesized that DUX4 is preferentially expressed only in affected muscles. Our newly generated DUX4p-GFP reporter mice grossly express GFP in areas that are preferentially affected in FSHD. In this Aim, we will perform a detailed characterization of GFP expression in our DUX4p-GFP mice. These results will help define the expected expression patterns of DUX4, and ultimately increase our understanding about the role of DUX4 FSHD pathogenesis. Specific Aim 2: To develop an AAV vector-mediated DUX4p-GFP mouse model. Previous endeavors to generate D4Z4 or DUX4 FSHD mouse models using traditional transgenic approaches have been unsuccessful. Although the previous attempts are not published in peer-reviewed literature, the difficulties encountered in generating these models were discussed in abstracts and talks at various scientific meetings over the last several years, including at the FSH Society’s 2008 International Research Consortium and Research Planning Meeting held in Philadelphia, Pennsylvania (https://www.fshdsociety.org/assets/pdf/FSHD_ASHG_IRC2008_Philadelphia_11Nov_ProgramAndAbstract s_proof.pdf). Vascular delivery of AAV vectors carrying FSHD-permissive D4Z4 repeats to adult mice may circumvent the early embryonic death or developmental defects arising from germline transmission of D4Z4 repeats using traditional methods. In this Aim, we will test the feasibility of using AAV vectors to drive D4Z4-specific expression patterns in mouse muscle using an AAV.DUX4p-GFP reporter vector. +
Screening FSHD patient-derivedmyoblasts, control myoblast, and muscle samples for expression changes at the proteomic level produced an unknown 50 kDa polypeptide highly expressed in FSHD samples compared to controls. Interestingly, this polypeptide is equally expressed in both normal and FSHD-patient derived myoblasts and early myotubes, however, unlike in control cells where its expression decreases, this unknown polypeptide remains highly expressed in differentiated muscle suggesting it is developmentally regulated and this regulation is disrupted in FSHD. This proposal will utilize standard biochemical techniques including column chromatography and mass spectrometry to purify and identify this 50 kDa putative FSHD biomarker. Subsequently, specific antibodies will be gerneated and characterized for further use to screen FSHD-derived cells to establish the universality of this biomarker. In addition, regardless of what its eventually identification turns out to be, identifying this protein will provide insight into FSHD pathophysiology, will be a useful FSHD biomarker, and may be one of the first proteins consistently and specifically upregulated in viable FSHD muscle. Therefore, generating specific and standardized antibodies to this protein will provide a useful resource for clinicians and basic FSHD researchers. +
The pathogenesis of FSHD has remained a mystery despite remarkable advances in the understanding of the underlying genetics. It was determined in 1992 that patients with FSHD have unusual contractions of a repeat element (so called D4Z4) at position 4q35 in the genome.1 However, what has remained elusive until now is how those contractions, i.e. loss of genomic material, could lead to an autosomal dominant disease. Within each D4Z4 repeat is a sequence termed Dux4 that encodes a putative double homeobox gene. Studies of the protein product have demonstrated that Dux4 overexpression can interfere with muscle differentiation. Thus, much effort has gone into the exploration of how D4Z4 repeats could lead to a “toxic-gain-of-function” related to the Dux4 transcript and protein. To date, no hypothesis has withstood experimental scrutiny. For one thing, there are individuals with D4Z4 contractions that do not develop FSHD. Recently, the group of van der Maarel reported in the journal Science their findings of the high resolution haplotype mapping of patients and unaffected individuals with D4Z4 contractions.2 Their findings provide evidence that the disease develops in individuals who have BOTH a D4Z4 repeat contraction AND a specific sequence in the pLAM domain at the 3’ end of the D4Z4 array (Figure 1). The D4Z4 repeat contraction results in “relaxed chromatin”, and allows the transcription of the Dux4 gene in the final D4Z4 repeat. However, it is the sequence in the pLAM domain that creates a site that is recognized by the cellular machinery allowing cleavage of the mRNA and the addition of a poly(A) tail. Without a poly(A) tail in the 3’ untranslated region (3’ UTR), transcripts are rapidly degraded and never translated into proteins.3 With these tails, transcripts are stabilized and appropriately localized in the cell, allowing for protein translation. In individuals who have D4Z4 contractions but a single base change in the distal sequence, the cell does not recognized it as a “polyadenylation signal” (PAS) site, no poly(A) tail is added to the 3’UTR of the transcript, the Dux4 transcript is unstable, no Dux4 protein is made, and the individuals are protected from getting the disease (Figure 1). Within this cascade are several opportunities, at least theoretically, to treat or even prevent FSHD in susceptible individuals. Any intervention that prevents the addition of the poly(A) tail to the Dux4 transcript is a potential therapeutic approach for FSHD. These findings suggest a direct line to a novel therapeutic approach. The toxicity leading to FSHD depends of effective mRNA processing in which the Dux4 transcript is cleaved and modified by the addition of a poly(A) tail. If one of these processes could be blocked, then the mRNA would be destabilized and the FSHD genotype would yield a normal phenotype. Clearly, it is untenable to interfere with mRNA processing in general because of the toxicity to the cell. Therefore, understanding the mechanisms by which a cell can bypass a specific PAS site would suggest a mechanism for selectively blocking the PAS site in the pLAM domain in the Dux4 gene without generally affecting cellular mRNA processing. This would be an effective treatment for patients with FSHD. +
Fascioscapulohumeral dystrophy (FSHD) is the third most common type of muscular dystrophy, with an estimated prevalence of 1 in 15,000 to 20,000 (Kissel, 1999) (Flanigan et al., 2001). It is an autosomal dominant disorder due to a deletion within the D4Z4 repeat region located on the subtelomeric region of chromosome 4q35. FSHD causes progressive atrophy and frequently asymmetrical weakness involving the face, shoulder girdle, upper arm, abdominal, and lower limb muscles. Most affected individuals develop symptoms during their second or third decade, with 20% eventually become wheelchair dependent (Padberg, Lunt, Koch & Fardeau, 1991) (Zatz et al., 1998). Early childhood onset of FSHD may be associated with more severe weakness as well as extra-neuromuscular manifestations such as mental retardation, retinal vasculopathy, and sensorineural hearing loss (Jardine et al., 1994) (Klinge et al., 2006). Although the majority of cases of FSHD are inherited, about 20%—30% of sporadic cases may occur as a result of spontaneous mutation or mosaicism (van der Maarel & Frants, 2005). Despite recent advances in the understanding of the molecular genetics of FSHD, the exact mechanism responsible for the disease remains unknown, and presently there is no cure (Tawil & Van Der Maarel, 2006) (van der Maarel, Frants & Padberg, 2007). As well, the prevalence, clinical variability, cross cultural presentation, and the psychosocial impact of FSHD on affected individuals constitute a significant public health concern. Emerging therapeutic trials will benefit from the availability of natural history data and reliable outcome measures (Rose & Tawil, 2004) (Tawil, 2008) for both children and adults with FSHD. Purpose of Study The main objectives of this study are: 1) to establish a standardized muscle testing protocol for use in children and youth with FSHD; 2) to describe the clinical phenotypes of pediatric onset FSHD; 3) to evaluate the impact of FSHD on health-related quality of life and disability across different age groups; and 4) to explore potential genetic modifiers of clinical phenotypes and disease progression in FSHD.
Archive
Download FSH Society grants awarded August 2018 cycle
Download FSH Society grants awarded February 2018 cycle
Download FSH Society grants awarded August 2017 cycle
Download FSH Society CTRN grant awarded August 2017
Download FSH Society grants awarded February 2017 cycle
Download FSH Society grants awarded August 2016 cycle
Download FSH Society grants awarded February 2016 cycle
Download FSH Society grants awarded August 2015 cycle
Download FSH Society grants awarded February 2015 & Ad Hoc 2015
Download FSH Society grants awarded from 2014 cycles
Download FSH Society grants awarded from February 2013 to February 2014 grant cycles
Download FSH Society grants awarded from 2011 and 2012 grant cycles (MS-Word)
Download FSH Society grants awarded from August 2009 and 2010 grant cycles (MS-Word)
Download FSH Society grants awarded in 2008
Download FSH Society grants awarded from 1998 to 2008 cycles (MS-Word)